
 Mонографията „Наследствени ретинални дистрофии” на доц. д-р Елена Мермеклиева описва същността на наследствените ретинални дистрофии, които са генетични заболявания от групата на „редките очни болести“. Разгледани са подробно етиопатогенезата, клиничната картина, съвременните диагностични методи за доказването им, както и възможностите и перспективите за лечение на отделните нозологични единици. Представена е най-актуалната научна информация по темата, както и интересни примери от практиката на автора. Разгледани са и социалните аспекти, свързани с тези редки, но силно инвалидизиращи индивида заболявания.Темата би била интересна за офталмолози, генетици, електрофизиолози, молекулярни биолози, оптометристи и социални работници, тъй като добрата грижа за тези пациенти изисква тясна колаборация и добро сътрудничество между всички тези специалисти.
Mонографията „Наследствени ретинални дистрофии” на доц. д-р Елена Мермеклиева описва същността на наследствените ретинални дистрофии, които са генетични заболявания от групата на „редките очни болести“. Разгледани са подробно етиопатогенезата, клиничната картина, съвременните диагностични методи за доказването им, както и възможностите и перспективите за лечение на отделните нозологични единици. Представена е най-актуалната научна информация по темата, както и интересни примери от практиката на автора. Разгледани са и социалните аспекти, свързани с тези редки, но силно инвалидизиращи индивида заболявания.Темата би била интересна за офталмолози, генетици, електрофизиолози, молекулярни биолози, оптометристи и социални работници, тъй като добрата грижа за тези пациенти изисква тясна колаборация и добро сътрудничество между всички тези специалисти.
 Дефицитът на алфа-1 антитрипсин (AATD) е рядко и недостатъчно добре диагностицирано състояние, което е свързано с развитието на чернодробно заболяване при възрастни и деца и белодробен емфизем при възрастни. Няколко проучвания показват, че познанията за болестта и нейната диагностика са ограничени сред специалистите по здравни грижи и че има значителна неравнопоставеност в достъпа до специализирани грижи и подходящо лечение в цяла Европа. Европейската комисия и Европейското респираторно общество (ERS) препоръчват грижите за пациенти с AATD да бъдат организирани в референтни центрове на национално или регионално ниво. Тези референтни центрове трябва да осигуряват оптимални клинични грижи по отношение на адекватни диагностични техники, като фенотипизиране и генотипизиране, и да осигуряват достъп до лечение съгласно указанията. Референтните центрове също трябва да осигуряват постоянно медицинско образование за медицинските специалисти, генетично консултиране, сътрудничество с асоциации на пациенти, както и да насърчават съвместни изследвания и клинични изпитвания с нови и съществуващи терапии за болестта. Тези центрове трябва да имат регистър на своята дейност и да си сътрудничат с големи, международни, многоцентрови регистри, като например международния регистър на Европейско сътрудничество за изследване на дефицита на Alpha-1 антитрипсин (EARCO), който е одобрен от ERS и има за цел да набере и проследи до 3000 пациенти за период от три години, за по-добро разбиране естествената история на заболяването и въздействието на различни лечения върху резултатите в реалния живот. Международното сътрудничество и стандартизираното събиране на висококачествени перспективни данни ще осигурят нови наблюдения върху клиничните прояви и прогнозата на AATD. Повече информация може да получите тук.
Дефицитът на алфа-1 антитрипсин (AATD) е рядко и недостатъчно добре диагностицирано състояние, което е свързано с развитието на чернодробно заболяване при възрастни и деца и белодробен емфизем при възрастни. Няколко проучвания показват, че познанията за болестта и нейната диагностика са ограничени сред специалистите по здравни грижи и че има значителна неравнопоставеност в достъпа до специализирани грижи и подходящо лечение в цяла Европа. Европейската комисия и Европейското респираторно общество (ERS) препоръчват грижите за пациенти с AATD да бъдат организирани в референтни центрове на национално или регионално ниво. Тези референтни центрове трябва да осигуряват оптимални клинични грижи по отношение на адекватни диагностични техники, като фенотипизиране и генотипизиране, и да осигуряват достъп до лечение съгласно указанията. Референтните центрове също трябва да осигуряват постоянно медицинско образование за медицинските специалисти, генетично консултиране, сътрудничество с асоциации на пациенти, както и да насърчават съвместни изследвания и клинични изпитвания с нови и съществуващи терапии за болестта. Тези центрове трябва да имат регистър на своята дейност и да си сътрудничат с големи, международни, многоцентрови регистри, като например международния регистър на Европейско сътрудничество за изследване на дефицита на Alpha-1 антитрипсин (EARCO), който е одобрен от ERS и има за цел да набере и проследи до 3000 пациенти за период от три години, за по-добро разбиране естествената история на заболяването и въздействието на различни лечения върху резултатите в реалния живот. Международното сътрудничество и стандартизираното събиране на висококачествени перспективни данни ще осигурят нови наблюдения върху клиничните прояви и прогнозата на AATD. Повече информация може да получите тук.
![]()
Видеото обяснява на пациентите, засегнати от редки, слабо разпространени и сложни заболявания, какво представляват Европейските Референтни Мрежи (ЕРМ) и как те биха могли да ги подкрепят при определяне на диагноза или лечение, в случай че техният медицински специалист счита, че е необходима подкрепата на ЕРМ. Видеото е достъпно на български език на уебсайта на Европейската комисия: https://europa.eu/!kP76Cq
„Предизвикателствата пред пациентите с вродена надбъбречна хиперплазия и техните лекари и родители” е една от темите, които бяха разгледани по време на XI Национална конференция за редки болести и лекарства сираци, проведена на 11 – 12 септември 2020 във Виртуалния конгресен център на Институт по редки болести. Лекцията e представена от проф. Виолета Йотова, директор на Експертния център по редки ендокринни болести във Варна и г-жа Вера Гледачева, член на Национален алианс на хората с редки болести. Гледайте цялото видео в YouTube канала на Институт по редки болести.
 Антитяло-зависимата клетъчна цитотоксичност (ADCC) е основният механизъм на действие за няколко предлагани на пазара терапевтични антитела (mAbs) и за много други включени в клинични изпитвания. Ефикасността на ADCC силно зависи от способността на терапевтичните mAbs да набират ефекторни клетки като естествени клетки убийци, които индуцират апоптозата на целевите клетки. Набирането на ефекторни клетки от mAbs се влияе отрицателно от модификация на фукоза на N-гликани върху Fc; по този начин, използването на афукозилиран mAbs е тенденция за подобрена ADCC терапия. Повечето афукозилирани mAbs в клинично или търговско производство са произведени от клетки гостоприемници на клетки от яйчници на китайски хамстер (CHO), като обикновено генерират ниски добиви в сравнение с дивизионен тип гостоприемник. Изследването описва подробно създаването и характеризирането на две модифицирани CHOZN® клетъчни линии, в които ензимът, участващ в синтеза на гуанозин дифосфат (GDP) -фукоза, GDP маноза-4,6-дехидратаза (Gmds) и GDP-L-фукоза синтаза (FX) ), е елиминиран. Най-добрите клетъчни линии на гостоприемника за всеки от нокаутите, FX – / – и Gmds – / -, са избрани въз основа на устойчивостта на растежа, избирателния толеранс към количеството MSX, производствения титър, нивото на фукозилиране и клетъчната стабилност. Тествано е производството на две патентовани IgG1 mAbs в модифицирани клетки гостоприемници и е установено, че титрите са сравними с клетките CHOZN®. MAbs, генерирани от която и да е от клетъчна линия на KO, показват липса на модификация на фукоза, което води до значително подсилени FcγRIIIa и ADCC ефекти. Тези данни демонстрират, че и двата вида клетки гостоприемници FX – / – и Gmds – / – могат да заменят клетки Fut8 – / – CHO за клинично производство на терапевтични антитела. Повече информация може да получите тук.
Антитяло-зависимата клетъчна цитотоксичност (ADCC) е основният механизъм на действие за няколко предлагани на пазара терапевтични антитела (mAbs) и за много други включени в клинични изпитвания. Ефикасността на ADCC силно зависи от способността на терапевтичните mAbs да набират ефекторни клетки като естествени клетки убийци, които индуцират апоптозата на целевите клетки. Набирането на ефекторни клетки от mAbs се влияе отрицателно от модификация на фукоза на N-гликани върху Fc; по този начин, използването на афукозилиран mAbs е тенденция за подобрена ADCC терапия. Повечето афукозилирани mAbs в клинично или търговско производство са произведени от клетки гостоприемници на клетки от яйчници на китайски хамстер (CHO), като обикновено генерират ниски добиви в сравнение с дивизионен тип гостоприемник. Изследването описва подробно създаването и характеризирането на две модифицирани CHOZN® клетъчни линии, в които ензимът, участващ в синтеза на гуанозин дифосфат (GDP) -фукоза, GDP маноза-4,6-дехидратаза (Gmds) и GDP-L-фукоза синтаза (FX) ), е елиминиран. Най-добрите клетъчни линии на гостоприемника за всеки от нокаутите, FX – / – и Gmds – / -, са избрани въз основа на устойчивостта на растежа, избирателния толеранс към количеството MSX, производствения титър, нивото на фукозилиране и клетъчната стабилност. Тествано е производството на две патентовани IgG1 mAbs в модифицирани клетки гостоприемници и е установено, че титрите са сравними с клетките CHOZN®. MAbs, генерирани от която и да е от клетъчна линия на KO, показват липса на модификация на фукоза, което води до значително подсилени FcγRIIIa и ADCC ефекти. Тези данни демонстрират, че и двата вида клетки гостоприемници FX – / – и Gmds – / – могат да заменят клетки Fut8 – / – CHO за клинично производство на терапевтични антитела. Повече информация може да получите тук.
 Множественият дефицит на сулфатаза (MSD) е ултра-рядко невродегенеративно разстройство, причинено от патогенни варианти на SUMF1. Този ген кодира генериращ формилглицин ензим (FGE), протеин необходим за активиране на сулфатазата. Клиничният ход на MSD е резултат от адитивния ефект на всеки дефицит на сулфатаза, включително метахроматична левкодистрофия (MLD), няколко мукополизахаридози (MPS II, IIIA, IIID, IIIE, IVA, VI), хондродисплазия и X-свързана ихтиоза. Известно е, че засегнатите индивиди демонстрират сложен и тежък фенотип, връзката генотип-фенотип и подробният клиничен ход са неизвестни. Докладвано е за 35 случая, включени в ретроспективно проучване за естествената история. Неврологичната функция е оценена с ретроспективни скали. Извършено е биохимично и математическо моделиране на нови варианти на SUMF1. Генотипите са класифицирани въз основа на прогнозираната функционална промяна и на всеки индивид е поставена стойност за тежест на генотипа. Средната възраст за поява на симптома е 0,25 години; средната възраст за поставяне на диагнозата е 2,7 години; средната възраст, при която настъпва фатален изход е 13 години. При всички индивиди е наблюдавано забавяне в развитието, а само при част от тях се стига до амбулация и вербална комуникация. Всички пациенти показват системно увеличаване на тежестта на симптомите. По-ранната възраст за поява на симптомите и тежките варианти на патогенност корелира с лоши неврологични резултати. Чрез използване на ретроспективно фенотипизиране и подробен анализ на вариантите е определена естествената история на MSD. Установено е, че атенюираните случаи могат да бъдат разграничени от тежките случаи по възраст на поява, постигане на амбулация и генотип. Резултатите от проучването могат да помогнат за изготвянето на прогнозата и да улеснят бъдещия дизайн на изследването. Повече информация може да получите тук.
Множественият дефицит на сулфатаза (MSD) е ултра-рядко невродегенеративно разстройство, причинено от патогенни варианти на SUMF1. Този ген кодира генериращ формилглицин ензим (FGE), протеин необходим за активиране на сулфатазата. Клиничният ход на MSD е резултат от адитивния ефект на всеки дефицит на сулфатаза, включително метахроматична левкодистрофия (MLD), няколко мукополизахаридози (MPS II, IIIA, IIID, IIIE, IVA, VI), хондродисплазия и X-свързана ихтиоза. Известно е, че засегнатите индивиди демонстрират сложен и тежък фенотип, връзката генотип-фенотип и подробният клиничен ход са неизвестни. Докладвано е за 35 случая, включени в ретроспективно проучване за естествената история. Неврологичната функция е оценена с ретроспективни скали. Извършено е биохимично и математическо моделиране на нови варианти на SUMF1. Генотипите са класифицирани въз основа на прогнозираната функционална промяна и на всеки индивид е поставена стойност за тежест на генотипа. Средната възраст за поява на симптома е 0,25 години; средната възраст за поставяне на диагнозата е 2,7 години; средната възраст, при която настъпва фатален изход е 13 години. При всички индивиди е наблюдавано забавяне в развитието, а само при част от тях се стига до амбулация и вербална комуникация. Всички пациенти показват системно увеличаване на тежестта на симптомите. По-ранната възраст за поява на симптомите и тежките варианти на патогенност корелира с лоши неврологични резултати. Чрез използване на ретроспективно фенотипизиране и подробен анализ на вариантите е определена естествената история на MSD. Установено е, че атенюираните случаи могат да бъдат разграничени от тежките случаи по възраст на поява, постигане на амбулация и генотип. Резултатите от проучването могат да помогнат за изготвянето на прогнозата и да улеснят бъдещия дизайн на изследването. Повече информация може да получите тук.
 Интересът към разработването на лекарства за редки заболявания се е увеличил драстично след приемането на Закона за лекарствата сираци в САЩ през 1983 г., като 40% от новите одобрения за лекарства през 2019 г. са насочени към редките симптоми. Ограниченото разбиране на естествената история и прогресирането на заболяването възпрепятства напредъка и увеличава рисковете, свързани с разработването на лекарства за редки заболявания. Използването на международни стандарти може да помогне за съгласуване на данните, да позволи техния обмен и обединяване в по-големи набори от данни и разбиране на естествената история на заболяването. Американската Агенцията за контрол на храните и лекарствата (FDA) изисква използването на Консорциум за стандарти за обмен на клинични данни (CDISC) при приемане на нови лекарства, за да се помогне на агенцията ефективно да получава, обработва, рецензира и архивира подадените документи, както и да помогне за обединяването на данните, за да отговори на изследователски въпроси. Такива бази данни са в основата на усилията за откриване на биомаркери и подходящи за целта модели, одобрени от регулаторите. В обзора е описано развитието на CDISC фармако-терапевтични ръководства за мускулна дистрофия на Дюшен и болест на Хънтингтън чрез консорциумите на Critical Path Institute. Тези ръководства описват официални структури от данни и стандартизирана терминология за картографиране и обединяване на информация от различни източници. Това води до по-добра стандартизация за събирането на данни и позволява обединяване и сравняване на данни от множество проучвания. Обединяването на тези данни дава възможност за по-добро разбиране на прогресията на заболяването, което може да помогне за преодоляване на често срещаните предизвикателства при дизайн на клинични изпитвания при тези и други редки заболявания. В обобщение, стандартизацията на клиничните данни ще доведе до по-бърз път към регулаторно одобрение на спешно необходими нови терапии за пациенти. Повече информация може да получите тук.
Интересът към разработването на лекарства за редки заболявания се е увеличил драстично след приемането на Закона за лекарствата сираци в САЩ през 1983 г., като 40% от новите одобрения за лекарства през 2019 г. са насочени към редките симптоми. Ограниченото разбиране на естествената история и прогресирането на заболяването възпрепятства напредъка и увеличава рисковете, свързани с разработването на лекарства за редки заболявания. Използването на международни стандарти може да помогне за съгласуване на данните, да позволи техния обмен и обединяване в по-големи набори от данни и разбиране на естествената история на заболяването. Американската Агенцията за контрол на храните и лекарствата (FDA) изисква използването на Консорциум за стандарти за обмен на клинични данни (CDISC) при приемане на нови лекарства, за да се помогне на агенцията ефективно да получава, обработва, рецензира и архивира подадените документи, както и да помогне за обединяването на данните, за да отговори на изследователски въпроси. Такива бази данни са в основата на усилията за откриване на биомаркери и подходящи за целта модели, одобрени от регулаторите. В обзора е описано развитието на CDISC фармако-терапевтични ръководства за мускулна дистрофия на Дюшен и болест на Хънтингтън чрез консорциумите на Critical Path Institute. Тези ръководства описват официални структури от данни и стандартизирана терминология за картографиране и обединяване на информация от различни източници. Това води до по-добра стандартизация за събирането на данни и позволява обединяване и сравняване на данни от множество проучвания. Обединяването на тези данни дава възможност за по-добро разбиране на прогресията на заболяването, което може да помогне за преодоляване на често срещаните предизвикателства при дизайн на клинични изпитвания при тези и други редки заболявания. В обобщение, стандартизацията на клиничните данни ще доведе до по-бърз път към регулаторно одобрение на спешно необходими нови терапии за пациенти. Повече информация може да получите тук.
 На 3 октомври от 16:00 ч. до 19:30 ч. ще се проведе онлайн събитието на Световен алианс на организациите за заболявания на хипофизата (WAPO), в който членува Асоциация Хипофиза. Събитието ще има превод на руски и английски език. Регистрацията е безплатна. Повече информация може да получите тук.
На 3 октомври от 16:00 ч. до 19:30 ч. ще се проведе онлайн събитието на Световен алианс на организациите за заболявания на хипофизата (WAPO), в който членува Асоциация Хипофиза. Събитието ще има превод на руски и английски език. Регистрацията е безплатна. Повече информация може да получите тук.
 Наследствените полиневропатии са разнородна група заболявания на периферната нервна система. В това проучване са изследвани демографските, клиничните, електрофизиологичните и генетичните характеристики на пациентите с наследствена полиневропатия, диагностицирани и проследявани в клиника в Измир, Турция. Пациентите, които са диагностицирани с наследствени полиневропатии по време на изследвания на нервната проводимост в клиниката, са оценени ретроспективно. В общо 1484 проучвания на нервната проводимост 207 пациенти са диагностицирани с полиневропатия. Деветдесет и девет от тези пациенти са установени, че имат наследствена полиневропатия, 52 от които са мъже и 47 са жени. Шестдесет и девет пациенти с наследствена полиневропатия отговарят на аксонална, а 30 на демиелинизираща полиневропатия. Генетичен анализ е извършен при 69 пациенти и 49 от тях са генетично диагностицирани, което е довело до степен на диагностика от 71%. Напредъкът в генетиката е довел до увеличаване на хетерогенността на наследствените полиневропатии, което води до затруднения при използването на съществуващите класификации. Въпреки че типичните мутации, които се очакват при възникналите в детството полиневропатии, се наблюдават по-рядко, полиневропатиите често се срещат като открития на сложни, мултисистемни заболявания. Повече информация може да получите тук.
Наследствените полиневропатии са разнородна група заболявания на периферната нервна система. В това проучване са изследвани демографските, клиничните, електрофизиологичните и генетичните характеристики на пациентите с наследствена полиневропатия, диагностицирани и проследявани в клиника в Измир, Турция. Пациентите, които са диагностицирани с наследствени полиневропатии по време на изследвания на нервната проводимост в клиниката, са оценени ретроспективно. В общо 1484 проучвания на нервната проводимост 207 пациенти са диагностицирани с полиневропатия. Деветдесет и девет от тези пациенти са установени, че имат наследствена полиневропатия, 52 от които са мъже и 47 са жени. Шестдесет и девет пациенти с наследствена полиневропатия отговарят на аксонална, а 30 на демиелинизираща полиневропатия. Генетичен анализ е извършен при 69 пациенти и 49 от тях са генетично диагностицирани, което е довело до степен на диагностика от 71%. Напредъкът в генетиката е довел до увеличаване на хетерогенността на наследствените полиневропатии, което води до затруднения при използването на съществуващите класификации. Въпреки че типичните мутации, които се очакват при възникналите в детството полиневропатии, се наблюдават по-рядко, полиневропатиите често се срещат като открития на сложни, мултисистемни заболявания. Повече информация може да получите тук.
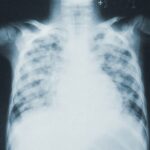 Саркоидозата е грануломатозно системно заболяване с неизвестна етиология, при което белият дроб е най-често засегнатият орган. Терапевтичното справяне със заболяването е предизвикателство, тъй като клиничното представяне и прогнозата са много разнородни. В настоящия обзор са обобщени основните постижения в терапията на саркоидозата. Текущите терапии на заболяването са категоризирани в три линии: глюкокортикоиди (първа линия), имуносупресори (втора линия) и биологични препарати (трета линия). Последните проучвания за глюкокортикоидите съобщават, че ефикасността може да бъде сходна при високи и ниски дози, но с увеличаване на страничните ефекти при по-високите дози. При имуносупресорите последните публикации, свързани с микофенолат и инжектиране на хранилище кортикотропин (RCI) добавят повече информация за тяхната употреба и ефикасност. Публикувани са нови доказателства за използването на анти–тумор некротичен фактор (анти-TNFα) при рефрактерна сърдечна саркоидоза и невросаркоидоза. Повече информация може да получите тук.
Саркоидозата е грануломатозно системно заболяване с неизвестна етиология, при което белият дроб е най-често засегнатият орган. Терапевтичното справяне със заболяването е предизвикателство, тъй като клиничното представяне и прогнозата са много разнородни. В настоящия обзор са обобщени основните постижения в терапията на саркоидозата. Текущите терапии на заболяването са категоризирани в три линии: глюкокортикоиди (първа линия), имуносупресори (втора линия) и биологични препарати (трета линия). Последните проучвания за глюкокортикоидите съобщават, че ефикасността може да бъде сходна при високи и ниски дози, но с увеличаване на страничните ефекти при по-високите дози. При имуносупресорите последните публикации, свързани с микофенолат и инжектиране на хранилище кортикотропин (RCI) добавят повече информация за тяхната употреба и ефикасност. Публикувани са нови доказателства за използването на анти–тумор некротичен фактор (анти-TNFα) при рефрактерна сърдечна саркоидоза и невросаркоидоза. Повече информация може да получите тук.