
 Оценката на иновацията, оттам и оценката на стойността на новите лекарствени терапии многократно са били обект на коментар в „Редки болести и лекарства сираци”. Това, разбира се, е ключов момент от оценката на здравните технологии, който често бива използван от индустрията, за да оправдае високата цена на съответния лекарствен продукт. Иновацията сама по себе си е широко понятие. Говорейки за стойност и оценка на здравните технологии обаче, важно измерение на иновацията на дадена терапия са нейните добавени ползи. Не ползи, а добавени ползи. Това вече е свързващото звено с ценообразуването, оттам и разходите за лечение с въпросната здравна технология. Повече информация може да получите тук.
Оценката на иновацията, оттам и оценката на стойността на новите лекарствени терапии многократно са били обект на коментар в „Редки болести и лекарства сираци”. Това, разбира се, е ключов момент от оценката на здравните технологии, който често бива използван от индустрията, за да оправдае високата цена на съответния лекарствен продукт. Иновацията сама по себе си е широко понятие. Говорейки за стойност и оценка на здравните технологии обаче, важно измерение на иновацията на дадена терапия са нейните добавени ползи. Не ползи, а добавени ползи. Това вече е свързващото звено с ценообразуването, оттам и разходите за лечение с въпросната здравна технология. Повече информация може да получите тук.
Вече е онлайн новият брой на нашето научно списание „Редки болести и лекарства сираци”. Изданието съдържа 4 публикации на различна тематика. Редакционната статия е „Иновация, добавени ползи и ценообразуване при оценката на здравни технологии”. Ако желаете да получите най-новата информация за редките болести и лекарства сираци, как се осъществява диагностиката и лечението в България, моля последвайте линка.
 Васкулитите са група от редки заболявания с различна проява и изход. Новите терапевтични възможности водят до необходимостта от дългосрочни регистри. Португалският регистър на ревматичните заболявания (Reuma.pt) е уеб базиран електронен запис на историята на заболяването, създаден през 2008 г., който понастоящем включва специфични модули за 12 заболявания и повече от 20 000 пациенти, регистрирани в 79 ревматологични центъра. През октомври 2014 г. е създаден специален модул за васкулитите като част от мрежата на Европейската лига против ревматизма, позволяващ събиране и централно съхранение на данните от пациенти с тези състояния. Събрани са данни, свързани с демографията, диагностиката, критериите за класификация, инструментите за оценка и лечение. Целта на обзора е да опише структурата на Reuma.pt/vasculitis и да характеризира пациентите, регистрирани от неговото създаване. Повече информация може да получите тук.
Васкулитите са група от редки заболявания с различна проява и изход. Новите терапевтични възможности водят до необходимостта от дългосрочни регистри. Португалският регистър на ревматичните заболявания (Reuma.pt) е уеб базиран електронен запис на историята на заболяването, създаден през 2008 г., който понастоящем включва специфични модули за 12 заболявания и повече от 20 000 пациенти, регистрирани в 79 ревматологични центъра. През октомври 2014 г. е създаден специален модул за васкулитите като част от мрежата на Европейската лига против ревматизма, позволяващ събиране и централно съхранение на данните от пациенти с тези състояния. Събрани са данни, свързани с демографията, диагностиката, критериите за класификация, инструментите за оценка и лечение. Целта на обзора е да опише структурата на Reuma.pt/vasculitis и да характеризира пациентите, регистрирани от неговото създаване. Повече информация може да получите тук.
 Синдромът на Ушер тип 1 (USH1) е рядко заболяване и основна причина за генетична глухо-слепота. Глухотата е налице от раждането, докато ретинитис пигментоза (RP), която е прогресираща, обикновено се проявява през детството и води до слепота. Целта на това изследване е да се разработи модел на заболяването, описващ симптомите при USH1 и тяхното въздействие върху живота на пациентите. Проведени са интервюта с пациенти (деца и възрастни), както и с родители на деца и юноши с USH1. Интервюираните пациенти са изследвани от офталмолози от специализирани очни центрове в САЩ и Франция. Обучените интервюиращи са използвали полуструктурирани техники, за да извлекат важни за пациентите и техните родители понятия. Тематичният анализ на записи от интервюта води до идентифициране на концепции, които са използвани за генериране на модел на заболяването. Повече информация може да получите тук.
Синдромът на Ушер тип 1 (USH1) е рядко заболяване и основна причина за генетична глухо-слепота. Глухотата е налице от раждането, докато ретинитис пигментоза (RP), която е прогресираща, обикновено се проявява през детството и води до слепота. Целта на това изследване е да се разработи модел на заболяването, описващ симптомите при USH1 и тяхното въздействие върху живота на пациентите. Проведени са интервюта с пациенти (деца и възрастни), както и с родители на деца и юноши с USH1. Интервюираните пациенти са изследвани от офталмолози от специализирани очни центрове в САЩ и Франция. Обучените интервюиращи са използвали полуструктурирани техники, за да извлекат важни за пациентите и техните родители понятия. Тематичният анализ на записи от интервюта води до идентифициране на концепции, които са използвани за генериране на модел на заболяването. Повече информация може да получите тук.
 Целоцентезата е инвазивна техника, която може да осигури пренатална диагностика на единични генни разстройства още от седма гестационна седмица. Целта на този обзор е да се проучи безопасността на целоцентезата. Извършена е целоцентеза за пренатална диагностика на хемоглобинопатии при 402 едноплодни бременности, при които и двамата родители са носители на белези на таласемия или на сърповидноклетъчна анемия. Направена е оценка за всяка майка на свързания с процедурата дискомфорт и болка, успеха при вземане на проби и получаване на резултати, на резултатите от бременността и следродилното проследяване. Повече информация може да получите тук.
Целоцентезата е инвазивна техника, която може да осигури пренатална диагностика на единични генни разстройства още от седма гестационна седмица. Целта на този обзор е да се проучи безопасността на целоцентезата. Извършена е целоцентеза за пренатална диагностика на хемоглобинопатии при 402 едноплодни бременности, при които и двамата родители са носители на белези на таласемия или на сърповидноклетъчна анемия. Направена е оценка за всяка майка на свързания с процедурата дискомфорт и болка, успеха при вземане на проби и получаване на резултати, на резултатите от бременността и следродилното проследяване. Повече информация може да получите тук.
 Повечето пациенти с алфа-1 антитрипсинов дефицит остават недиагностицирани и следователно не могат да се възползват от настоящите терапии. На 23 юни 2019 г. в Орландо, Флорида се е провел семинар, на който заинтересованите страни от изследователските, фармацевтичните и пациентските общности са се фокусирали върху темата за откриване на алфа-1 антитрипсинов дефицит. Таргетните групи са пациенти с хронична обструктивна белодробна болест, необяснимо хронично чернодробно заболяване, както и членове от семейството на засегнати лица.Скринингът на новородените, извличането на данни от електронни медицински записи и осигуряване на възможност за лабораторна диагностика остават опции за разработване на бъдещи стратегии за ранно откриване. Повече информация може да получите тук.
Повечето пациенти с алфа-1 антитрипсинов дефицит остават недиагностицирани и следователно не могат да се възползват от настоящите терапии. На 23 юни 2019 г. в Орландо, Флорида се е провел семинар, на който заинтересованите страни от изследователските, фармацевтичните и пациентските общности са се фокусирали върху темата за откриване на алфа-1 антитрипсинов дефицит. Таргетните групи са пациенти с хронична обструктивна белодробна болест, необяснимо хронично чернодробно заболяване, както и членове от семейството на засегнати лица.Скринингът на новородените, извличането на данни от електронни медицински записи и осигуряване на възможност за лабораторна диагностика остават опции за разработване на бъдещи стратегии за ранно откриване. Повече информация може да получите тук.
 Митохондриалните разстройства са група от редки заболявания, причинени от ядрени или митохондриални мутации в ДНК. Тяхната подчертана клинична и генетична хетерогенност, както и препоръките за установяване на отклонения правят трудно оценяването на разпространението на базата на фенотипа. Обзора показва изчисленията на риска за продължителност на живота за всички известни автозомно-рецесивни митохондриални разстройства въз основа на генетични данни. Въз основа на публично достъпната база данни за агрегиране на генома (gnomAD) и вътрешна база данни за екзома е оценена алелната честота на гените на причиняващите заболявания, свързани с автозомни рецесивни митохондриални разстройства. Спрямо това е оценен риска за продължителност на живота при 249 автозомно-рецесивни митохондриални разстройства. Повече информация може да получите тук.
Митохондриалните разстройства са група от редки заболявания, причинени от ядрени или митохондриални мутации в ДНК. Тяхната подчертана клинична и генетична хетерогенност, както и препоръките за установяване на отклонения правят трудно оценяването на разпространението на базата на фенотипа. Обзора показва изчисленията на риска за продължителност на живота за всички известни автозомно-рецесивни митохондриални разстройства въз основа на генетични данни. Въз основа на публично достъпната база данни за агрегиране на генома (gnomAD) и вътрешна база данни за екзома е оценена алелната честота на гените на причиняващите заболявания, свързани с автозомни рецесивни митохондриални разстройства. Спрямо това е оценен риска за продължителност на живота при 249 автозомно-рецесивни митохондриални разстройства. Повече информация може да получите тук.
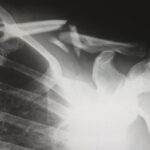 Х-свързаният хипофосфатемичен (XLH) рахит е рядко заболяване, често неправилно диагностицирано и третирано. Въпреки че има клинични указания, които предлагат някои терапевтични препоръки въз основа на клиничния опит на експерти, лекарите все още имат въпроси относно някои важни аспекти при диагностицирането и лечението на XLH, като например кога трябва да подозираме заболяването, кой трябва да отговаря за диагностицирането, какво трябва да се направи след диагностициране на заболяването или какви терапевтични възможности са налични в момента. Целта на този обзор е да отговори на някои от по-честите въпроси, свързани с лечението на пациенти с XLH от група експерти, участващи в научна конференция за XLH в Мадрид през 2019 г.. Повече информация може да получите тук.
Х-свързаният хипофосфатемичен (XLH) рахит е рядко заболяване, често неправилно диагностицирано и третирано. Въпреки че има клинични указания, които предлагат някои терапевтични препоръки въз основа на клиничния опит на експерти, лекарите все още имат въпроси относно някои важни аспекти при диагностицирането и лечението на XLH, като например кога трябва да подозираме заболяването, кой трябва да отговаря за диагностицирането, какво трябва да се направи след диагностициране на заболяването или какви терапевтични възможности са налични в момента. Целта на този обзор е да отговори на някои от по-честите въпроси, свързани с лечението на пациенти с XLH от група експерти, участващи в научна конференция за XLH в Мадрид през 2019 г.. Повече информация може да получите тук.
 Сирингомиелията и синдромът на Киари са класифицирани като редки болести, но понастоящем липсват данни за честотата на тези заболявания в Европа. Повишената способност за диагностициране на тези патологии чрез магнитен резонанс и неговата широка достъпност водят до увеличаване на докладваните случаи, често безсимптомни, с необходимостта от стандартизиране на дефиниции, диагностични критерии и лечение. Целите на това изследване са да представят наличните междурегионални препоръки, разработени с основната цел оценяване разпространението и честотата на сирингомиелията и синдрома на Киари в Северозападна Италия, със специално внимание върху симптоматичните форми. Повече информация може да получите тук.
Сирингомиелията и синдромът на Киари са класифицирани като редки болести, но понастоящем липсват данни за честотата на тези заболявания в Европа. Повишената способност за диагностициране на тези патологии чрез магнитен резонанс и неговата широка достъпност водят до увеличаване на докладваните случаи, често безсимптомни, с необходимостта от стандартизиране на дефиниции, диагностични критерии и лечение. Целите на това изследване са да представят наличните междурегионални препоръки, разработени с основната цел оценяване разпространението и честотата на сирингомиелията и синдрома на Киари в Северозападна Италия, със специално внимание върху симптоматичните форми. Повече информация може да получите тук.
 Наследствените спастични параплегии (НСП) са хетерогенна група от редки невродегенеративни нарушения, които се проявяват със спастичност на долните крайници. Заболяването е известно като сложна НСП, ако спастичността е придружена от допълнителни характеристики като когнитивно увреждане, малкомозъчен синдром, оскъден корпус калозум или невропатия. Повечето семейства с НСП показват автозомно-доминантен тип унаследяване. От друга страна, случаите на автозомно-рецесивен тип унаследяване са често срещани, поради високата честота на междуродствените бракове. Този обзор има за цел да проучи клиничната и генетична етиология при група пациенти с НСП. Повече информация може да получите тук.
Наследствените спастични параплегии (НСП) са хетерогенна група от редки невродегенеративни нарушения, които се проявяват със спастичност на долните крайници. Заболяването е известно като сложна НСП, ако спастичността е придружена от допълнителни характеристики като когнитивно увреждане, малкомозъчен синдром, оскъден корпус калозум или невропатия. Повечето семейства с НСП показват автозомно-доминантен тип унаследяване. От друга страна, случаите на автозомно-рецесивен тип унаследяване са често срещани, поради високата честота на междуродствените бракове. Този обзор има за цел да проучи клиничната и генетична етиология при група пациенти с НСП. Повече информация може да получите тук.