
 The assessment of innovation and the value of new medicinal therapies, has been repeatedly commented in “Rare Diseases and Orphan Drugs” journal. This is a key point in health technology assessment, which is often used by industry to justify the high cost of a particular drug. Innovation itself is a broad concept. However, when it comes to the value and assessment of health technologies, an important dimension of the innovation of a therapy is its added benefits. Not benefits, but added benefits. This is the link between pricing and cost of treatment with the health technology in question. For more information click here.
The assessment of innovation and the value of new medicinal therapies, has been repeatedly commented in “Rare Diseases and Orphan Drugs” journal. This is a key point in health technology assessment, which is often used by industry to justify the high cost of a particular drug. Innovation itself is a broad concept. However, when it comes to the value and assessment of health technologies, an important dimension of the innovation of a therapy is its added benefits. Not benefits, but added benefits. This is the link between pricing and cost of treatment with the health technology in question. For more information click here.
The new issue of our scientific journal Rare Diseases and Orphan Drugs is now online. The issue contains 4 publications on various topics. The editorial is “Innovation, added benefits and pricing in health technology assessment”. If you would like to receive the latest information on rare diseases and orphan drugs, how the diagnostics and treatment is performed Bulgaria, please follow the link.
 The vasculitides are a group of rare diseases with different manifestations and outcomes. New therapeutic options have led to the need for long-term registries. The Rheumatic Diseases Portuguese Register, Reuma.pt, is a web-based electronic clinical record, created in 2008, which currently includes specific modules for 12 diseases and > 20,000 patients registered from 79 rheumatology centres. On October 2014, a dedicated module for vasculitis was created as part of the European Vasculitis Society collaborative network, enabling prospective collection and central storage of encrypted data from patients with this condition. All Portuguese rheumatology centres were invited to participate. Data regarding demographics, diagnosis, classification criteria, assessment tools, and treatment were collected. We aim to describe the structure of Reuma.pt/vasculitis and characterize the patients registered since its development. For more information click here.
The vasculitides are a group of rare diseases with different manifestations and outcomes. New therapeutic options have led to the need for long-term registries. The Rheumatic Diseases Portuguese Register, Reuma.pt, is a web-based electronic clinical record, created in 2008, which currently includes specific modules for 12 diseases and > 20,000 patients registered from 79 rheumatology centres. On October 2014, a dedicated module for vasculitis was created as part of the European Vasculitis Society collaborative network, enabling prospective collection and central storage of encrypted data from patients with this condition. All Portuguese rheumatology centres were invited to participate. Data regarding demographics, diagnosis, classification criteria, assessment tools, and treatment were collected. We aim to describe the structure of Reuma.pt/vasculitis and characterize the patients registered since its development. For more information click here.
 Type 1 Usher syndrome (USH1) is a rare disease and major cause of genetic deaf-blindness. Deafness is present from birth while retinitis pigmentosa (RP) which typically presents during childhood is progressive leading to blindness. The aim of this research was to develop a disease model describing USH1 symptoms and their impact on patients’ lives. Qualitative interviews were conducted with patients (pediatric and adult) and parents of children and adolescents with USH1. Interviewed subjects were enrolled through ophthalmologists from specialized eye centers in the USA and in France. Trained interviewers used semi-structured techniques to elicit concepts relevant to patients and their parents. Thematic analysis of interview transcripts led to the identification of concepts which were organized to generate a disease model. For more information click here.
Type 1 Usher syndrome (USH1) is a rare disease and major cause of genetic deaf-blindness. Deafness is present from birth while retinitis pigmentosa (RP) which typically presents during childhood is progressive leading to blindness. The aim of this research was to develop a disease model describing USH1 symptoms and their impact on patients’ lives. Qualitative interviews were conducted with patients (pediatric and adult) and parents of children and adolescents with USH1. Interviewed subjects were enrolled through ophthalmologists from specialized eye centers in the USA and in France. Trained interviewers used semi-structured techniques to elicit concepts relevant to patients and their parents. Thematic analysis of interview transcripts led to the identification of concepts which were organized to generate a disease model. For more information click here.
 Celocentesis, is an invasive technique that can provide prenatal diagnosis of single gene disorders, from as early as seven weeks’ gestation. The objective of this study is to examine the safety of celocentesis. Celocentesis was performed for prenatal diagnosis of hemoglobinopathies in 402 singleton pregnancies, in which both parents were carriers of thalassaemia or sickle cell disease trait. We assessed procedure-related maternal discomfort or pain, success of sampling and obtaining of results, pregnancy outcome and postnatal follow up. For more information click here.
Celocentesis, is an invasive technique that can provide prenatal diagnosis of single gene disorders, from as early as seven weeks’ gestation. The objective of this study is to examine the safety of celocentesis. Celocentesis was performed for prenatal diagnosis of hemoglobinopathies in 402 singleton pregnancies, in which both parents were carriers of thalassaemia or sickle cell disease trait. We assessed procedure-related maternal discomfort or pain, success of sampling and obtaining of results, pregnancy outcome and postnatal follow up. For more information click here.
 Most patients with alpha-1 antitrypsin deficiency remain undiagnosed and therefore do not benefit from current therapies or become eligible for research studies of new treatments under development. Improving the detection rate for AATD is therefore a high priority for the Alpha-1 Foundation. A workshop was held on June 23, 2019 in Orlando, Florida during which stakeholders from the research, pharmaceutical, and patient communities focused on the topic of alpha-1 antitrypsin deficiency detection. A variety of detection strategies have been explored in the past and new approaches are emerging as technology advances. Targeted detection includes patients with chronic obstructive pulmonary disease, unexplained chronic liver disease, and family members of affected individuals. Newborn screening, electronic medical record data mining, and direct-to-consumer testing remain options for future detection strategies. For more information click here.
Most patients with alpha-1 antitrypsin deficiency remain undiagnosed and therefore do not benefit from current therapies or become eligible for research studies of new treatments under development. Improving the detection rate for AATD is therefore a high priority for the Alpha-1 Foundation. A workshop was held on June 23, 2019 in Orlando, Florida during which stakeholders from the research, pharmaceutical, and patient communities focused on the topic of alpha-1 antitrypsin deficiency detection. A variety of detection strategies have been explored in the past and new approaches are emerging as technology advances. Targeted detection includes patients with chronic obstructive pulmonary disease, unexplained chronic liver disease, and family members of affected individuals. Newborn screening, electronic medical record data mining, and direct-to-consumer testing remain options for future detection strategies. For more information click here.
 Mitochondrial disorders are a group of rare diseases, caused by nuclear or mitochondrial DNA mutations. Their marked clinical and genetic heterogeneity as well as referral and ascertainment biases render phenotype-based prevalence estimations difficult. Here we calculated the lifetime risk of all known autosomal recessive mitochondrial disorders on basis of genetic data. We queried the publicly available Genome Aggregation Database (gnomAD) and our in-house exome database to assess the allele frequency of disease-causing variants in genes associated with autosomal recessive mitochondrial disorders. Based on this, we estimated the lifetime risk of 249 autosomal recessive mitochondrial disorders. Three of these disorders and phenylketonuria (PKU) served as a proof of concept since calculations could be aligned with known birth prevalence data from newborn screening reports. For more information click here.
Mitochondrial disorders are a group of rare diseases, caused by nuclear or mitochondrial DNA mutations. Their marked clinical and genetic heterogeneity as well as referral and ascertainment biases render phenotype-based prevalence estimations difficult. Here we calculated the lifetime risk of all known autosomal recessive mitochondrial disorders on basis of genetic data. We queried the publicly available Genome Aggregation Database (gnomAD) and our in-house exome database to assess the allele frequency of disease-causing variants in genes associated with autosomal recessive mitochondrial disorders. Based on this, we estimated the lifetime risk of 249 autosomal recessive mitochondrial disorders. Three of these disorders and phenylketonuria (PKU) served as a proof of concept since calculations could be aligned with known birth prevalence data from newborn screening reports. For more information click here.
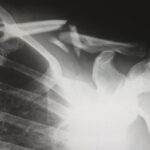 X-linked hypophosphataemia (XLH) rickets is a rare disease frequently misdiagnosed and mismanaged. Despite having clinical guidelines that offers some therapeutic recommendations based on the clinical experience of experts, physicians still have questions about some important aspects of the diagnosis and treatment of XLH, such as when the disease should be suspected, who should be in charge of the diagnosis, what should be done once the disease is diagnosed, or what therapeutic options are currently available. The objective of this paper is to answer some of the more frequent questions related to the management of patients with XLH by a group of experts participating in a scientific conference on XLH held in Madrid. For more information click here.
X-linked hypophosphataemia (XLH) rickets is a rare disease frequently misdiagnosed and mismanaged. Despite having clinical guidelines that offers some therapeutic recommendations based on the clinical experience of experts, physicians still have questions about some important aspects of the diagnosis and treatment of XLH, such as when the disease should be suspected, who should be in charge of the diagnosis, what should be done once the disease is diagnosed, or what therapeutic options are currently available. The objective of this paper is to answer some of the more frequent questions related to the management of patients with XLH by a group of experts participating in a scientific conference on XLH held in Madrid. For more information click here.
 Syringomyelia and Chiari Syndrome are classified as rare diseases, but current known occurrence in Europe is missing. The increased ability to diagnose these pathologies by magnetic resonance imaging and its widespread availability has led to an increase of reported cases, often asymptomatic, with the need to standardize definitions, diagnostic criteria and treatments. The aims of this study are to present shared Interregional Recommendations developed with the primary aim to estimate Syringomyelia and Chiari Syndrome prevalence and incidence in North Western Italy, with special reference to symptomatic forms. For more information click here.
Syringomyelia and Chiari Syndrome are classified as rare diseases, but current known occurrence in Europe is missing. The increased ability to diagnose these pathologies by magnetic resonance imaging and its widespread availability has led to an increase of reported cases, often asymptomatic, with the need to standardize definitions, diagnostic criteria and treatments. The aims of this study are to present shared Interregional Recommendations developed with the primary aim to estimate Syringomyelia and Chiari Syndrome prevalence and incidence in North Western Italy, with special reference to symptomatic forms. For more information click here.
 Hereditary spastic paraplegias (HSPs) are a heterogenous group of rare neurodegenerative disorders that present with lower limb spasticity. It is known as complicated HSP if spasticity is accompanied by additional features such as cognitive impairment, cerebellar syndrome, thin corpus callosum, or neuropathy. Most HSP families show autosomal dominant (AD) inheritance. On the other hand, autosomal recessive (AR) cases are also common because of the high frequency of consanguineous marriages in our country. This study aimed to investigate the clinical and genetic aetiology in a group of HSP patients. For more information click here.
Hereditary spastic paraplegias (HSPs) are a heterogenous group of rare neurodegenerative disorders that present with lower limb spasticity. It is known as complicated HSP if spasticity is accompanied by additional features such as cognitive impairment, cerebellar syndrome, thin corpus callosum, or neuropathy. Most HSP families show autosomal dominant (AD) inheritance. On the other hand, autosomal recessive (AR) cases are also common because of the high frequency of consanguineous marriages in our country. This study aimed to investigate the clinical and genetic aetiology in a group of HSP patients. For more information click here.