
 Diagnostic Exome Sequencing yields a single genetic diagnosis in approximately 30% of cases, and according to recent studies the prevalence of identifying two genetic conditions in a single individual ranges between 4.6 and 7%. We present a patient diagnosed with three different rare conditions, each explained by a pathogenic mutation in a different gene. A 17 year old female was evaluated for history of motor and speech delay, scoliosis, distinctive craniofacial features and dry skin in the Department of Clinical Genomics at Mayo Clinic. Previous diagnostic testing was unrevealing including biochemical testing, echocardiogram, abdominal ultrasound and electroencephalogram. For more information click here.
Diagnostic Exome Sequencing yields a single genetic diagnosis in approximately 30% of cases, and according to recent studies the prevalence of identifying two genetic conditions in a single individual ranges between 4.6 and 7%. We present a patient diagnosed with three different rare conditions, each explained by a pathogenic mutation in a different gene. A 17 year old female was evaluated for history of motor and speech delay, scoliosis, distinctive craniofacial features and dry skin in the Department of Clinical Genomics at Mayo Clinic. Previous diagnostic testing was unrevealing including biochemical testing, echocardiogram, abdominal ultrasound and electroencephalogram. For more information click here.
 Peripheral neuropathy, organomegaly, endocrinopathy, monoclonal protein and skin changes (POEMS) syndrome is a rare disease, and only in a minority of cases, causes an impairment of kidney function. Here, we describe a case of a 55-year-old man with a history of POEMS syndrome who presented with acute kidney injury following a routine blood test. On further investigation, a relapse in POEMS syndrome was diagnosed, uniquely isolated to renal involvement. For more information click here.
Peripheral neuropathy, organomegaly, endocrinopathy, monoclonal protein and skin changes (POEMS) syndrome is a rare disease, and only in a minority of cases, causes an impairment of kidney function. Here, we describe a case of a 55-year-old man with a history of POEMS syndrome who presented with acute kidney injury following a routine blood test. On further investigation, a relapse in POEMS syndrome was diagnosed, uniquely isolated to renal involvement. For more information click here.
 The European commission published a document with the last uptate of Frequently Asked Questions (FAQ) and Specific criteria for the ERNs call for membership. FAQ include the most common questions like: what is an ERN; what is the role of the Board of MS; are only rare diseases included in the scope of ERNs; How to apply to become a member of an ERNs; how will applications be assessed; what does the conflict of interest policy cover and many more. For more information click here.
The European commission published a document with the last uptate of Frequently Asked Questions (FAQ) and Specific criteria for the ERNs call for membership. FAQ include the most common questions like: what is an ERN; what is the role of the Board of MS; are only rare diseases included in the scope of ERNs; How to apply to become a member of an ERNs; how will applications be assessed; what does the conflict of interest policy cover and many more. For more information click here.
 Current American Academy of Pediatrics guidelines for children with Down syndrome (DS) recommend a complete blood count (CBC) at birth and hemoglobin annually to screen for iron deficiency (ID) and ID anemia (IDA) in low-risk children. We aimed to determine if macrocytosis masks the diagnosis of ID/IDA and to evaluate the utility of biochemical and red blood cell indices for detecting ID/IDA in DS. We reviewed data from 856 individuals from five DS specialty clinics. Data included hemoglobin, mean corpuscular volume, red cell distribution width (RDW), percent transferrin saturation (TS), ferritin, and c-reactive protein. Receiver operating characteristic curves were calculated. Macrocytosis was found in 32% of the sample. If hemoglobin alone was used for screening, all individuals with IDA would have been identified, but ID would have been missed in all subjects. RDW had the highest discriminability of any single test for ID/IDA. The combination of RDW with ferritin or TS led to 100% sensitivity, and RDW combined with ferritin showed the highest discriminability for ID/IDA. We provide evidence to support that a CBC and ferritin be obtained routinely for children over 1 year old with DS rather than hemoglobin alone for detection of ID. For more information click here.
Current American Academy of Pediatrics guidelines for children with Down syndrome (DS) recommend a complete blood count (CBC) at birth and hemoglobin annually to screen for iron deficiency (ID) and ID anemia (IDA) in low-risk children. We aimed to determine if macrocytosis masks the diagnosis of ID/IDA and to evaluate the utility of biochemical and red blood cell indices for detecting ID/IDA in DS. We reviewed data from 856 individuals from five DS specialty clinics. Data included hemoglobin, mean corpuscular volume, red cell distribution width (RDW), percent transferrin saturation (TS), ferritin, and c-reactive protein. Receiver operating characteristic curves were calculated. Macrocytosis was found in 32% of the sample. If hemoglobin alone was used for screening, all individuals with IDA would have been identified, but ID would have been missed in all subjects. RDW had the highest discriminability of any single test for ID/IDA. The combination of RDW with ferritin or TS led to 100% sensitivity, and RDW combined with ferritin showed the highest discriminability for ID/IDA. We provide evidence to support that a CBC and ferritin be obtained routinely for children over 1 year old with DS rather than hemoglobin alone for detection of ID. For more information click here.
European Reference Networks (ERNs) were created under the 2011 Directive on Patient Rights’ in Cross-Border Healthcare. They are based on the voluntary cooperation of Member States that contribute to the ERNs’ activities in accordance with their national legislation. ERNs are operational since March 2017. To ensure a proper and sustainable functioning of the ERNs and to reap all benefits for patients suffering from rare and low prevalence complex diseases across the EU, the ERNs need to be linked in a clear and stable way to the healthcare systems of the Member States. These are issues of key importance and demand tangible actions. This Statement aims to give incentives to Member States to further enhance the integration process based on the input provided by the Working Group on Integration. Member States are encouraged to facilitate the integration of ERNs to their healthcare systems by: assessing and if needed adapting or updating the national policy and/or legal framework; creating appropriate (clear and well-defined) patients’ pathways in order to improve the care and management of patients with complex or rare diseases; developing clear systems for referral to ERNs; developing a clear strategy for communicating and disseminating information about ERNs; reflecting on the means to best support. For more information click here.
 The EURORDIS Photo Award is one of the 14 Black Pearl Awards that recognise the stars of the rare disease community, as nominated by the rare disease community and members of the public. Whether you are an amateur photographer or took that one great photo that illustrates the challenges or joy of living with a rare disease, you can submit your photo by 16 January to be in with a chance of winning the EURORDIS Photo Award 2020! For more information click here.
The EURORDIS Photo Award is one of the 14 Black Pearl Awards that recognise the stars of the rare disease community, as nominated by the rare disease community and members of the public. Whether you are an amateur photographer or took that one great photo that illustrates the challenges or joy of living with a rare disease, you can submit your photo by 16 January to be in with a chance of winning the EURORDIS Photo Award 2020! For more information click here.
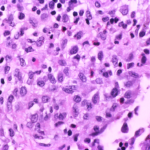 Langerhans cell histiocytosis (LCH) is a rare disease of unknown aetiology. While it may affect any organ of the body, few cases of solitary lung involvement are published in the literature. Here, we report a rare case of pulmonary LCH (PLCH) in an adult. A 52-year-old male presented to hospital in July 2018 with complaints of progressively worsening cough with sputum, breathlessness, easy fatigability, and loss of appetite since 2016, and a 32-year history of heavy cigarette smoking (average 30 cigarettes/d). Physical examination showed only weakened breathing sounds and wheezing during lung auscultation. Chest computed tomography (CT) showed irregular micronodules and multiple thin-walled small holes. Respiratory function tests showed a slight decrease. Ultrasonic cardiogram showed mild tricuspid regurgitation and no pulmonary hypertension. Fibreoptic bronchoscopy was performed with transbronchial biopsies from the basal segment of right lower lobe. LCH was confirmed by immunohistochemistry. The final diagnosis was PLCH without extra-pulmonary involvement. We suggested smoking cessation treatment. A 3-mo follow-up chest CT scan showed clear absorption of the nodule and thin-walled small holes. The symptoms of cough and phlegm had improved markedly and appetite had improved. There was no obvious dyspnoea. Imaging manifestations of nodules, cavitating nodules, and thick-walled or thin-walled cysts prompted suspicion of PLCH and lung biopsy for diagnosis. For more information click here.
Langerhans cell histiocytosis (LCH) is a rare disease of unknown aetiology. While it may affect any organ of the body, few cases of solitary lung involvement are published in the literature. Here, we report a rare case of pulmonary LCH (PLCH) in an adult. A 52-year-old male presented to hospital in July 2018 with complaints of progressively worsening cough with sputum, breathlessness, easy fatigability, and loss of appetite since 2016, and a 32-year history of heavy cigarette smoking (average 30 cigarettes/d). Physical examination showed only weakened breathing sounds and wheezing during lung auscultation. Chest computed tomography (CT) showed irregular micronodules and multiple thin-walled small holes. Respiratory function tests showed a slight decrease. Ultrasonic cardiogram showed mild tricuspid regurgitation and no pulmonary hypertension. Fibreoptic bronchoscopy was performed with transbronchial biopsies from the basal segment of right lower lobe. LCH was confirmed by immunohistochemistry. The final diagnosis was PLCH without extra-pulmonary involvement. We suggested smoking cessation treatment. A 3-mo follow-up chest CT scan showed clear absorption of the nodule and thin-walled small holes. The symptoms of cough and phlegm had improved markedly and appetite had improved. There was no obvious dyspnoea. Imaging manifestations of nodules, cavitating nodules, and thick-walled or thin-walled cysts prompted suspicion of PLCH and lung biopsy for diagnosis. For more information click here.
 Idiopathic pulmonary fibrosis (IPF) is a chronic pulmonary disease that is complicated by diagnostic challenges, multiple comorbidities, and a poor prognosis. Although considered a relatively rare disease, healthcare costs are substantial and disproportionate to the incidence and prevalence of the disease. The comorbidities associated with IPF not only complicate treatment strategies but also increase the burden for patients via higher costs and undesirable health outcomes. Historically, pharmacologic treatment options for IPF have been limited and are often associated with low efficacy. Two drugs approved for IPF, nintedanib and pirfenidone, offer promise for improving health outcomes and survival during the course of the disease. Considerations of cost and adverse events are important when planning treatment options. Optimizing care through patient-centered care management programs can improve outcomes and health-related quality of life for patients. Such programs emphasize communication between healthcare professionals and patients in order to educate patients on their condition, so they can make informed healthcare decisions. Disease registries can be important tools for optimizing data collection and analysis for a disease with limited incidence and prevalence. For more information click here.
Idiopathic pulmonary fibrosis (IPF) is a chronic pulmonary disease that is complicated by diagnostic challenges, multiple comorbidities, and a poor prognosis. Although considered a relatively rare disease, healthcare costs are substantial and disproportionate to the incidence and prevalence of the disease. The comorbidities associated with IPF not only complicate treatment strategies but also increase the burden for patients via higher costs and undesirable health outcomes. Historically, pharmacologic treatment options for IPF have been limited and are often associated with low efficacy. Two drugs approved for IPF, nintedanib and pirfenidone, offer promise for improving health outcomes and survival during the course of the disease. Considerations of cost and adverse events are important when planning treatment options. Optimizing care through patient-centered care management programs can improve outcomes and health-related quality of life for patients. Such programs emphasize communication between healthcare professionals and patients in order to educate patients on their condition, so they can make informed healthcare decisions. Disease registries can be important tools for optimizing data collection and analysis for a disease with limited incidence and prevalence. For more information click here.
 Rett syndrome (RTT) is an early-onset neurodevelopmental disorder that primarily affects females, resulting in severe cognitive and physical disabilities, and is one of the most prevalent causes of intellectual disability in females. More than fifty years after the first publication on Rett syndrome, and almost two decades since the first report linking RTT to the MECP2 gene, the research community’s effort is focused on obtaining a better understanding of the genetics and the complex biology of RTT and Rett-like phenotypes without MECP2 mutations. Herein, we review the current molecular genetic studies, which investigate the genetic causes of RTT or Rett-like phenotypes which overlap with other genetic disorders and document the swift evolution of the techniques and methodologies employed. This review also underlines the clinical and genetic heterogeneity of the Rett syndrome spectrum and provides an overview of the RTT-related genes described to date, many of which are involved in epigenetic gene regulation, neurotransmitter action or RNA transcription/translation. Finally, it discusses the importance of including both phenotypic and genetic diagnosis to provide proper genetic counselling from a patient’s perspective and the appropriate treatment. For more information click here.
Rett syndrome (RTT) is an early-onset neurodevelopmental disorder that primarily affects females, resulting in severe cognitive and physical disabilities, and is one of the most prevalent causes of intellectual disability in females. More than fifty years after the first publication on Rett syndrome, and almost two decades since the first report linking RTT to the MECP2 gene, the research community’s effort is focused on obtaining a better understanding of the genetics and the complex biology of RTT and Rett-like phenotypes without MECP2 mutations. Herein, we review the current molecular genetic studies, which investigate the genetic causes of RTT or Rett-like phenotypes which overlap with other genetic disorders and document the swift evolution of the techniques and methodologies employed. This review also underlines the clinical and genetic heterogeneity of the Rett syndrome spectrum and provides an overview of the RTT-related genes described to date, many of which are involved in epigenetic gene regulation, neurotransmitter action or RNA transcription/translation. Finally, it discusses the importance of including both phenotypic and genetic diagnosis to provide proper genetic counselling from a patient’s perspective and the appropriate treatment. For more information click here.
![]() Fabry disease and Pompe disease are rare lysosomal storage disorders that belong to a heterogeneous group of more than 200 distinct inborn metabolic diseases. Mutations followed by loss of function of enzymes or transporters that are localised in the acidic environment of the lysosome may result in degradation of many substrates, such as glycosaminoglycans, glycosphingolipids, glycogen, cholesterol, oligosaccharides, glycoproteins, and peptides, or the excretion of the products degraded by the lysosome. Our aim was to identify the oral signs and symptoms of Fabry disease and Pompe disease from a systematic review made using MEDLINE/PubMed, and a hand search for relevant articles, following the PRISMA guidelines. Both diseases show various craniofacial and oral changes, including supernumerary teeth, dental agenesis, angiokeratoma, and telangiectases in Fabry disease; and macroglossia, teeth fusion, and taurodontism in Pompe disease. Common clinical signs of Fabry disease include hyposalivation, hypohidrosis, and xerophthalmia, and a generally reduced physical resilience was apparent in patients with Pompe disease. Oral and craniofacial changes in patients with both diseases extend over their entire lifetime and can be detected even in an infant. Lysosomal storage diseases should be taken into consideration in the differential diagnosis of relevant diverse symptoms, because treatment, when available, is most effective when started early. The main therapeutic concepts are enzymatic replacement for Pompe disease, whereas patients with Fabry disease require additional oral chaperone treatment or enzyme replacement. For more information click here.
Fabry disease and Pompe disease are rare lysosomal storage disorders that belong to a heterogeneous group of more than 200 distinct inborn metabolic diseases. Mutations followed by loss of function of enzymes or transporters that are localised in the acidic environment of the lysosome may result in degradation of many substrates, such as glycosaminoglycans, glycosphingolipids, glycogen, cholesterol, oligosaccharides, glycoproteins, and peptides, or the excretion of the products degraded by the lysosome. Our aim was to identify the oral signs and symptoms of Fabry disease and Pompe disease from a systematic review made using MEDLINE/PubMed, and a hand search for relevant articles, following the PRISMA guidelines. Both diseases show various craniofacial and oral changes, including supernumerary teeth, dental agenesis, angiokeratoma, and telangiectases in Fabry disease; and macroglossia, teeth fusion, and taurodontism in Pompe disease. Common clinical signs of Fabry disease include hyposalivation, hypohidrosis, and xerophthalmia, and a generally reduced physical resilience was apparent in patients with Pompe disease. Oral and craniofacial changes in patients with both diseases extend over their entire lifetime and can be detected even in an infant. Lysosomal storage diseases should be taken into consideration in the differential diagnosis of relevant diverse symptoms, because treatment, when available, is most effective when started early. The main therapeutic concepts are enzymatic replacement for Pompe disease, whereas patients with Fabry disease require additional oral chaperone treatment or enzyme replacement. For more information click here.