
 В нашето списание “Редки болести и лекарства сираци” е публикувана нова статия на тема „Оценка на здравни технологии при целеви терапии“. Целта е да бъдат представени специфичните особености на оценката на здравни технологии (ОЗТ) при целеви терапии и да се обсъдят методологичните предизвикателства, свързани с приложението й в България. Направено е аналитично изследване на базата на литературен обзор и контент анализ на методологични ръководства за ОЗТ и на нормативната база у нас. Анализирани са особеностите и предизвикателствата на ОЗТ при целеви терапии въз основа на характеристиките на субекта и обекта на анализа. Дефинирани са субекта и обекта на ОЗТ и видовете целеви терапии. Дискутирани са особеностите и предизвикателствата пред ОЗТ при целеви терапии. Отделено е специално внимание на въпроса за съпътстващите диагностични изследвания при целевите терапии. Налице са съществени методологични особености на ОЗТ при целевите терапии, произтичащи от спецификата на обекта на анализа. Тези особености трябва да бъдат съблюдавани, както в процеса на изготвянето на докладите за ОЗТ, така и при вземането на здравнополитически решения, базирани на тези оценки. Повече информация може да получите тук.
В нашето списание “Редки болести и лекарства сираци” е публикувана нова статия на тема „Оценка на здравни технологии при целеви терапии“. Целта е да бъдат представени специфичните особености на оценката на здравни технологии (ОЗТ) при целеви терапии и да се обсъдят методологичните предизвикателства, свързани с приложението й в България. Направено е аналитично изследване на базата на литературен обзор и контент анализ на методологични ръководства за ОЗТ и на нормативната база у нас. Анализирани са особеностите и предизвикателствата на ОЗТ при целеви терапии въз основа на характеристиките на субекта и обекта на анализа. Дефинирани са субекта и обекта на ОЗТ и видовете целеви терапии. Дискутирани са особеностите и предизвикателствата пред ОЗТ при целеви терапии. Отделено е специално внимание на въпроса за съпътстващите диагностични изследвания при целевите терапии. Налице са съществени методологични особености на ОЗТ при целевите терапии, произтичащи от спецификата на обекта на анализа. Тези особености трябва да бъдат съблюдавани, както в процеса на изготвянето на докладите за ОЗТ, така и при вземането на здравнополитически решения, базирани на тези оценки. Повече информация може да получите тук.
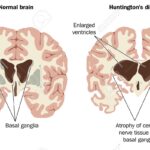 Болестта на Хънтингтън е рядко наследствено, невродегенеративно заболяване, което се предава автозомно-доминантно и засяга 10 на 100 000 души в западните част на света. Причината е мутация в гена на Хънтингтън, който съдържа ексцесивно количество от тринуклеотида CAG и е нестабилен и може да увеличи дължината си в следващите поколения. Като се има предвид непредвидимата възраст, в която започва развитието на заболяването и непредсказуемата клинична прогноза, точната интерпретация, подходяща психологична подкрепа и научно обоснована интерпретация на резултатите от генетичния тест са от решаващо значение в контекста на добрата клинична практика и при назначаването на терапия. Повече информация може да получите тук.
Болестта на Хънтингтън е рядко наследствено, невродегенеративно заболяване, което се предава автозомно-доминантно и засяга 10 на 100 000 души в западните част на света. Причината е мутация в гена на Хънтингтън, който съдържа ексцесивно количество от тринуклеотида CAG и е нестабилен и може да увеличи дължината си в следващите поколения. Като се има предвид непредвидимата възраст, в която започва развитието на заболяването и непредсказуемата клинична прогноза, точната интерпретация, подходяща психологична подкрепа и научно обоснована интерпретация на резултатите от генетичния тест са от решаващо значение в контекста на добрата клинична практика и при назначаването на терапия. Повече информация може да получите тук.
Преконцепционна употреба на фолиева киселина за първична профилактика на редки генетични заболявания
В нашето списание “Редки болести и лекарства сираци” е публикувана нова статия на тема „Преконцепционна употреба на фолиева киселина за първична профилактика на редки генетични заболявания“. Дефектите на невралната тръба (ДНТ) представляват група тежки вродени аномалии, които са свързани с висока смъртност, неблагоприятна прогноза и дългосрочни увреждания, както и със значителни емоционални, психологически и икономически последствия. Всяка година в ЕС се наблюдават приблизително 5 000 случаи на ДНТ. Повечето от тях се диагностицират пренатално с последващо прекратяване на бременността. Установено е, че преконцепционният прием на фолиева киселина може значително да намали риска за ДНТ. Въпреки това обаче, към днешна дата в европейските страни не съществуват програми и политики за прием на фолиева киселина при жени в репродуктивна възраст или по-специално при тези, които планират бременност. Настоящата публикация има за цел да представи и анализира новите тенденции при употребата на фолиева киселина като средство за първична профилактика на редки генетични заболявания. Повече информация може да получите тук.
СИНОНИМИ: Kohlschütter-Tönz syndrome (KTS)
КОД МКБ10: G40.8
КОД ORPHANET:ORPHA1946
ТАЗИ ИНФОРМАЦИЯ ВИ СЕ ПРЕДОСТАВЯ НАПЪЛНО БЕЗПЛАТНО С ОБРАЗОВАТЕЛНА ЦЕЛ И НЕ ТРЯБВА ДА СЛУЖИ ЗА САМОДИАГНОСТИКА И САМОЛЕЧЕНИЕ. ПРИ НАЛИЧИЕ НА ЗДРАВЕН ПРОБЛЕМ, СЛЕДВА ДА СЕ ОБЪРНЕТЕ КЪМ ЛИЧНИЯ/ЛЕКУВАЩ ЛЕКАР.
КРАТКА ДЕФИНИЦИЯ НА БОЛЕСТТА: Синдромът на Kohlschütter-Tönz (KTS) е генетично хетерогенен автозомно-рецесивен синдром, характеризиращ се с триада амелогенезис имперфекта, инфантилна начална епилепсия, интелектуални затруднения с или без регресия и деменция.
ЕТИОЛОГИЯ: Синдромът на Kohlschutter-Tonz (KTZS) се причинява от хомозиготна или съставна хетерозиготна мутация в ROGDI гена на хромозома 16р13. В последните години се доказа и участието на ген SLC13A5 в етиологията на заболяването.
КЛИНИЧНИ ПРОЯВИ: Най-често срещаните фенотипове и симптоми отнасящи се към този синдром включват нарушение на интелектуалното развитие, припадъци, цялостно забавяне на развитието, нисък ръст, микроцефалия, атаксия, спастичност, вентрикуломегалия.Според данни на Human phenotype ontology(HPO) симптомите, свързани с този синдром, се подреждат на база честота на проявление по следния начин:
- 80-99% от пациентите – амелогенезис имперфекта, деменция, забавено развитие, нарушения в ЕЕГ, забавяне в интелектуалното развитие, гърчове, спастичност, пожълтяване на зъбите.
- 30-79% от пациентите – хипохидрозис.
- 5-29% от пациентите – цялостно забавяне на развитието, хидроцефалия, нисък ръст.
ГЕНЕТИЧНА КОНСУЛТАЦИЯ: Докладите за засегнатите пациенти показват автозомно-рецесивно наследяване на синдрома на Kohlschutter-Tonz. Разпространение: <1/1 000 000.
ЛЕКАРСТВО СИРАК: –
АСОЦИАЦИИ НА ПАЦИЕНТИ:
НАЦИОНАЛЕН АЛИАНС НА ХОРАТА С РЕДКИ БОЛЕСТИ
Лице за контакти: Владимир Томов
Електронна поща: tomov@raredis.org
Адрес за кореспонденция: бул.“Драган Цанков“ бл. 59-63 вх. 7, София 1000
СПЕЦИАЛИЗИРАНИ ЛАБОРАТОРИИ:
Лаборатория по молекулярна патология, СБАЛАГ «Майчин дом», София
СПЕЦИАЛИЗИРАНИ КЛИНИКИ:
Център по нервно-мускулни заболявания, Клиника по неврология, Александровска болница, София
Медицински център РареДис
Специализиран медицински център за физиотерапия и рехабилитация, детски болести, генетични консултации.
Електронна поща: medical@raredis.org
Адрес за кореспонденция: гр. Пловдив, ЖК Тракия, ул. „Маестро Г. Атанасов“ 22
Телефон: (032) 575 797
Интернет сайт: medical.raredis.org
ДОПЪЛНИТЕЛНА ИНФОРМАЦИЯ:
За допълнителна информация, моля посетете сайтовете на Orphanet и NIH Office of Rare Diseases Research.
Ако желаете да се абонирате за безплатния електронен бюлетин “Редки болести & Лекарства сираци”, моля въведете Вашия e-mail тук.
Описанието е изготвено от Д-р Златин Иванов, МЦ „РареДис“ – Пловдив
–––––––––––––––––––––––––
Last modification: 12:40 21.05.2024
–––––––––––––––––––––––––
 През последното десетилетие са създадени няколко големи регистри, включващи хиляди пациенти с идиопатична пулмонална фиброза (ИПФ). Пациентите, проучени в тези регистри имат широк спектър от тежки съпътстващи заболявания и могат да бъдат проследени за по-дълъг период от време. Данните от регистрите спомагат за по-доброто разбиране на клиничните характеристики на пациентите с ИПФ, въздействието, което болестта оказва върху тяхното качество на живот и оцеляване, както и начинът на диагностика. В бъдеще анализите на биологични материали, които ще бъдат описани в медицинската документация на пациентите, ще дадат възможност за идентифициране на биомаркери, свързани с прогресирането на заболяването, улеснявайки развитието на точните лекарствени подходи за терапия и прогноза при пациенти с ИПФ. Повче информация може да получите тук.
През последното десетилетие са създадени няколко големи регистри, включващи хиляди пациенти с идиопатична пулмонална фиброза (ИПФ). Пациентите, проучени в тези регистри имат широк спектър от тежки съпътстващи заболявания и могат да бъдат проследени за по-дълъг период от време. Данните от регистрите спомагат за по-доброто разбиране на клиничните характеристики на пациентите с ИПФ, въздействието, което болестта оказва върху тяхното качество на живот и оцеляване, както и начинът на диагностика. В бъдеще анализите на биологични материали, които ще бъдат описани в медицинската документация на пациентите, ще дадат възможност за идентифициране на биомаркери, свързани с прогресирането на заболяването, улеснявайки развитието на точните лекарствени подходи за терапия и прогноза при пациенти с ИПФ. Повче информация може да получите тук.
В нашето списание “Редки болести и лекарства сираци” е публикувана нова статия на тема „Анализ на разходите за провеждане на интравенозна бифосфонатна терапия при пациенти с онкологични заболявания”. Публикуван е ретроспективен анализ на преките разходи за провеждане на интравенозна бифосфонатна (БФ) терапия за периода 2012 – 2016 г. Данните са извлечени от регистрите на аптечните бази на болничните заведения в област Пловдив, в които се прилага БФ терапия. В допълнение са анализирани и годишните разходи за золедронова киселина, достъпни от регистъра на НЗОК. Всички разходи са количествено определени на базата на национални ставки, определени от законодателя. Преките разходи за лекарствена терапия за страната спадат 32 пъти от 2018 г. до 2012 г. – от 8 983 283.42 лв. до 276 722.51 лв. Тази тенденция се наблюдава и при данните от болничните аптеки, при които спадът е 99 пъти от 2012 г. до 2016 г. – от 2 334 138.90 лв. до 23 542.65 лв, но тук влияние оказва не само спада в цената на терапията, но и близо 4 пъти по-малкия брой на заплатените опаковки – от 4004 бр. за 2012 до 1402 бр. за 2016 г. Важна последица от болестта е, че индивидите не могат да изпълняват обичайните си ежедневни дейности и така освен, че се разходва бюджета на НЗОК се търпят и производствени загуби. Повече информация може да получите тук.
 Диагностиката на наследствените заболявания на кръвта все още е предизвикателство, особено при заболявания на тромбоцитите, поради хетерогенността на клиничния и лабораторния фенотип, ограничената специфичност на тестовете за функцията на тромбоцитите и големия брой гени, които може да са причина за заболяването. Съвременните технологии за секвениране позволяват едновременно и бързо изследване на множество гени на минимална цена и включват секвениране на цял екзон или секвенциране на целия геном. Въпреки наличието на тези технологии, много пациенти все още не са диагностицирани. Тъй като наследствените заболявания на кръвта са сложни и редки, насърчава се разработването на по-съвременни лабораторни анализи, подобрения в биоинформатичните изследвания и формирането на мултидисциплинарни екипи. . Повече информация може да получите тук.
Диагностиката на наследствените заболявания на кръвта все още е предизвикателство, особено при заболявания на тромбоцитите, поради хетерогенността на клиничния и лабораторния фенотип, ограничената специфичност на тестовете за функцията на тромбоцитите и големия брой гени, които може да са причина за заболяването. Съвременните технологии за секвениране позволяват едновременно и бързо изследване на множество гени на минимална цена и включват секвениране на цял екзон или секвенциране на целия геном. Въпреки наличието на тези технологии, много пациенти все още не са диагностицирани. Тъй като наследствените заболявания на кръвта са сложни и редки, насърчава се разработването на по-съвременни лабораторни анализи, подобрения в биоинформатичните изследвания и формирането на мултидисциплинарни екипи. . Повече информация може да получите тук.
СИНОНИМИ: –
КОД МКБ10: –
КОД ORPHANET: ORPHA1452
ТАЗИ ИНФОРМАЦИЯ ВИ СЕ ПРЕДОСТАВЯ НАПЪЛНО БЕЗПЛАТНО С ОБРАЗОВАТЕЛНА ЦЕЛ И НЕ ТРЯБВА ДА СЛУЖИ ЗА САМОДИАГНОСТИКА И САМОЛЕЧЕНИЕ. ПРИ НАЛИЧИЕ НА ЗДРАВЕН ПРОБЛЕМ, СЛЕДВА ДА СЕ ОБЪРНЕТЕ КЪМ ЛИЧНИЯ/ЛЕКУВАЩ ЛЕКАР.
КРАТКА ДЕФИНИЦИЯ НА БОЛЕСТТА:Клейдокраниалната дисплазия (CCD) е рядка генетична аномалия на развитието на костите, характеризираща се с хипопластични или апластични ключици, персистенция на широко отворени шрифтове и шевове и многобройни зъбни аномалии.
ЕТИОЛОГИЯ: CCD се причинява от мутации в RUNX2 гена (6p21), участващи в диференциацията на остеобластите и образуването на кости. Идентифицирани са широк спектър от мутации с висока проницаемост и значителна вариабилност. Не са установени ясни корелации на фенотип-генотип.
КЛИНИЧНИ ПРОЯВИ:Има изключително широк спектър от клинични прояви (дори в рамките на едно и също семейство) от изолирани зъбни аномалии до тежки малформации с функционални последици. Основните клинични признаци са хипоплазия или аплазия на ключиците с тесни, наклонени рамене, които могат да бъдат приближени отпред, забавено сливане на краниални конци с големи, широко отворени шрифтове при раждане, които могат да продължат през целия живот и широк спектър от зъбни аномалии, включително анормални зъби, равномерни или хаотични свръх-брой зъби (хипердонтия) в първичната и вторичната зъби, водещи до струпване и неправилно прилепване, задържане на млечни зъби, забавено изригване на вторични зъби и неуспех при изхвърляне на първичните зъби. Денталните прояви могат да повлияят на артикулацията и дъвченето. Други признаци включват широко плоско чело, хипертелоризъм, хипоплазия на средната част на тялото и заострена челюст, която придава характерен облик на лицето, както и брахидактилия, изтъняващи пръсти и къси, широки палци. Асоциираните скелетни аномалии включват нисък ръст, сколиоза, genu valgum, pes planus, широка pubic symphysis, диспластична лопатка и coxa vara, обикновено с малко клинично значение. Вторичните усложнения включват рецидивиращи инфекции на горните дихателни пътища, сънна апнея, леко моторно забавяне и различна степен на загуба на слуха. Когнитивните и интелектуалните функции са нормални. Делът на жените с КБК, изискващи цезарово сечение, е по-висок, отколкото в общата популация поради цепно-мозъчна диспропорция.
ГЕНЕТИЧНА КОНСУЛТАЦИЯ:Клейдокраниалната дисплазия следва автозомно доминантния модел на наследяване. На засегнатите семейства трябва да се предоставят генетични консултации. Броят на случаите, свързани с мутациите de novo, изглежда висок.
ЛЕЧЕНИЕ:Лечението на зъбните аномалии е много важно с цел постигане на оптимална функция и естетика. Трябва да се обмисли стоматологична хирургия за необработените зъби и ортодонтията за прилепване. Може да се наложи терапия с логопед. Антибиотиците се препоръчват при повтарящи се инфекции. Поради ролята на RUNX2 в поддържането на костите и осификацията, трябва да се следи костната минерална плътност и да се обмисли превантивното лечение на остеопорозата.
АСОЦИАЦИИ НА ПАЦИЕНТИ:
НАЦИОНАЛЕН АЛИАНС НА ХОРАТА С РЕДКИ БОЛЕСТИ
Лице за контакти: Владимир Томов
Електронна поща: tomov@raredis.org
Адрес за кореспонденция: бул.“Драган Цанков“ бл. 59-63 вх. 7, София 1000
СПЕЦИАЛИЗИРАНИ КЛИНИКИ В БЪЛГАРИЯ:
Медицински център РареДис
Специализиран медицински център за физиотерапия и рехабилитация, детски болести, генетични консултации.
Електронна поща: medical@raredis.org
Адрес за кореспонденция: гр. Пловдив, ЖК Тракия, ул. „Маестро Г. Атанасов“ 22
Телефон: (032) 575 797
Интернет сайт: www.medical.raredis.org
ДОПЪЛНИТЕЛНА ИНФОРМАЦИЯ:
За допълнителна информация, моля посетете сайтовете на Orphanet и NIH Office of Rare Diseases Research.
Описанието е изготвено от д-р Златин Иванов, МЦ „РареДис“ – Пловдив

Tимидин киназа 2 (ТК2) е митохондриална киназа, която фосфорилира пиримидиновите нуклеозиди тимидин и дезоксицитидин. Рецесивните мутации в TK2 гена са отговорни за „миопатичната форма“ на синдрома на изчерпване на митохондриалната ДНК. Описани са 18 пациента с митохондриална миопатия, поради мутация в гена за ТК2, при които липсва симптоматика до 12 годишна възраст. Средната възраст за начало на симптоматиката е 31 години. Първият симптом е слабост на мускулите на крайниците, птоза на клепачите и дихателна недостатъчност. Всички пациенти развиват променлива мускулна слабост с прогресиране на заболяването и показват слабост на дихателната мускулатура с необходимост от неинвазивна механична вентилация. Половината от пациентите имат затруднения при преглъщане. Пациенти с късно начало на ТК2 дефицит имат постоянен и разпознаваем клиничен фенотип и лоша прогноза, поради високия риск от ранна и прогресивна дихателна недостатъчност. Повече информация може да получите тук.
 Извършено е проучване, сравняващо различните модели на съдово увреждане в зависимост от пола при пациенти с артериит на Такаясу. Събрани са ретроспективно данни от 117 диагностицирани пациенти (11 мъже и 106 жени). Сравнени са разликите между мъжете и жените по отношение на демографските особености, закъснението в диагностиката, признаците и симптомите, свързани със заболяването, както и разположението на засегнатите артерии. Данните са получени от три публикувани статии, описващи различията между половете в артериита на Такаясу. Забавянето в диагностиката е по-високо при мъжете. Пациентите от мъжки пол показват по-често засягане на илиачните артерии, жените по-често страдат от клаудикацио на горните крайници. При пациентите с артериит на Такаясу полът има силно влияние върху модела на съдово засягане и следователно върху клиничната картина. По-специално, жените имат по-голямо участие на наддиафрагмалните съдове, докато при мъжете по-често се засягат коремните съдове. Повече информация може да получите тук.
Извършено е проучване, сравняващо различните модели на съдово увреждане в зависимост от пола при пациенти с артериит на Такаясу. Събрани са ретроспективно данни от 117 диагностицирани пациенти (11 мъже и 106 жени). Сравнени са разликите между мъжете и жените по отношение на демографските особености, закъснението в диагностиката, признаците и симптомите, свързани със заболяването, както и разположението на засегнатите артерии. Данните са получени от три публикувани статии, описващи различията между половете в артериита на Такаясу. Забавянето в диагностиката е по-високо при мъжете. Пациентите от мъжки пол показват по-често засягане на илиачните артерии, жените по-често страдат от клаудикацио на горните крайници. При пациентите с артериит на Такаясу полът има силно влияние върху модела на съдово засягане и следователно върху клиничната картина. По-специално, жените имат по-голямо участие на наддиафрагмалните съдове, докато при мъжете по-често се засягат коремните съдове. Повече информация може да получите тук.