
 Fanconi-Bickel syndrome (FBS) is a rare autosomal recessive disorder characterized by impaired glucose and galactose utilization as well as proximal renal tubular dysfunction. In this study clinical, biochemical, genetic, treatment, and follow-up data for 11 pediatric patients with FBS are retrospectively analysed.
Fanconi-Bickel syndrome (FBS) is a rare autosomal recessive disorder characterized by impaired glucose and galactose utilization as well as proximal renal tubular dysfunction. In this study clinical, biochemical, genetic, treatment, and follow-up data for 11 pediatric patients with FBS are retrospectively analysed.
Treatment interventions, including uncooked cornstarch and conventional rickets therapy, are leading to the remission of hepatomegaly and significant improvements in health markers. Genetic analysis is revealing 14 variants of the SLC2A2 gene, including six novel ones, providing insights into potential genetic links. Patients with specific genetic variations are displaying varying degrees of metabolic acidosis.The study is also uncovering intriguing findings in nonconsanguineous families, where rare homozygous variations are associated with regions of homozygosity, suggesting a potential mechanism of uniparental disomy 3.
In conclusion, the researchers emphasize the ongoing challenge of early Fanconi-Bickel syndrome diagnosis due to diverse initial symptoms. They caution against using recombinant human growth hormone for patients with normal growth hormone stimulation tests and suggest alkali supplementation for those with specific genetic variations linked to urine bicarbonate loss. The ongoing discovery of regions of homozygosity is providing valuable insights into the mechanisms behind rare homozygous variants in nonconsanguineous families. Read the full article here.
Expediting rare disease diagnosis: a call to bridge the gap between clinical and functional genomics
 Approximately 400 million people throughout the world suffer from a rare disease. Although advances in whole exome and whole genome sequencing have greatly facilitated rare disease diagnosis, overall diagnostic rates remain below 50%. Furthermore, in cases where accurate diagnosis is achieved the process requires an average of 4.8 years. Reducing the time required for disease diagnosis is among the most critical needs of patients impacted by a rare disease.
Approximately 400 million people throughout the world suffer from a rare disease. Although advances in whole exome and whole genome sequencing have greatly facilitated rare disease diagnosis, overall diagnostic rates remain below 50%. Furthermore, in cases where accurate diagnosis is achieved the process requires an average of 4.8 years. Reducing the time required for disease diagnosis is among the most critical needs of patients impacted by a rare disease.
In this article current challenges associated with rare disease diagnosis are described and several cutting-edge functional genomic screening technologies that have the potential to rapidly accelerate the process of distinguishing pathogenic variants that lead to disease are discussed. Read the full article here.
 Georgi Iskrov, Vyara Angelova , Boyan Bochev, Vaska Valchinova, Teodora Gencheva, Desislava Dzhuleva, Julian Dichev, Tanya Nedkova, Mariya Palkova , Anelia Tyutyukova, Maria Hristova, Eleonora Hristova-Atanasova, and Rumen Stefanov, as part of the Institute for Rare Diseases, Department of Social Medicine and Public Health and Medical Faculty at the Medical University – Plovdiv conducted a comprehensive survey exploring the perspectives of specialists in pediatrics, neonatology, medical genetics, and biochemistry on expanding the range of diseases covered by universal newborn screening in Bulgaria.
Georgi Iskrov, Vyara Angelova , Boyan Bochev, Vaska Valchinova, Teodora Gencheva, Desislava Dzhuleva, Julian Dichev, Tanya Nedkova, Mariya Palkova , Anelia Tyutyukova, Maria Hristova, Eleonora Hristova-Atanasova, and Rumen Stefanov, as part of the Institute for Rare Diseases, Department of Social Medicine and Public Health and Medical Faculty at the Medical University – Plovdiv conducted a comprehensive survey exploring the perspectives of specialists in pediatrics, neonatology, medical genetics, and biochemistry on expanding the range of diseases covered by universal newborn screening in Bulgaria.
Current research study named “Prospects for Expansion of Universal Newborn Screening in Bulgaria: A Survey among Medical Professionals” has been published in the prestigious international journal “International Journal of Neonatal screening” (Impact factor: 3.5).
Defining the scope of a newborn screening program poses a complex challenge in health policy. This study aims to explore the viewpoints of specialists in pediatrics, neonatology, medical genetics, and biochemistry regarding the potential expansion of the disease panel for universal newborn screening in Bulgaria. Conducted as an online survey from March to May 2022, the questionnaire presented 35 disorders that could potentially be incorporated into the Bulgarian panel for universal newborn screening. Participants, when supporting a specific condition, were required to justify their stance by evaluating its alignment with the ten principles of Wilson and Jungner.
The results revealed a substantial understanding of the existing universal newborn screening program in Bulgaria, with an overwhelming majority (97.4%) expressing support for expanding the panel to include additional conditions. Notably, four disorders received more than 50% approval for inclusion: cystic fibrosis (87.0%), thalassemia (72.7%), spinal muscular atrophy (65.6%), and classical galactosemia (59.1%). The perception of a condition as a significant health issue emerged as the most influential factor in garnering support. Concerns primarily centered around the costs associated with diagnosis and treatment.
The findings of this study emphasize the need for country-specific economic evaluations and additional research involving various stakeholders, such as government entities, payers, and patient organizations, to comprehensively comprehend and navigate the intricate landscape of newborn screening policymaking. Read the full article here.
 Hereditary angioedema with normal C1-INH (HAE-nC1-INH) is a rare genetic disease with similar phenotype to HAE-C1-INH, but different genetic background. Currently six subtypes are recognized, based on the underlying mutations.
Hereditary angioedema with normal C1-INH (HAE-nC1-INH) is a rare genetic disease with similar phenotype to HAE-C1-INH, but different genetic background. Currently six subtypes are recognized, based on the underlying mutations.
The objective of this study is to assess clinical features of patients with genetically characterized HAE-nC1-INH, from the North of Portugal. To achieve this retrospective assessment of clinical data from all patients with HAE-nC1-INH followed at a Hereditary Angioedema (HAE) Reference Center is performed.
Forty-one patients are identified, four with no family history. The FXII-mutation Thr328Lys (38 carriers) is the most prevalent. Three new potentially disease causing variants linked to HAE-nC1-INH are identified (c.529+4A>G:FXII; Cys248*:Kininogen-1; Arg261His:Plasminogen).
In this study the first Portuguese series of patients with HAE-nC1-INH genetically characterized is described. Differences with others may contribute to improve current unmet needs and raise awareness of this rare disease. Alsothe identification of 3 new variants(additional molecular studies are ongoing) and the first report of erythema marginatum in HAE-n-C1INH is highlighted. Read the full article here.
 For the 15th consecutive year, the National Conference on Rare Diseases and Orphan Drugs is set to take place, organized by the Institute for Rare Diseases.
For the 15th consecutive year, the National Conference on Rare Diseases and Orphan Drugs is set to take place, organized by the Institute for Rare Diseases.
The conference is the most significant forum in the field of rare diseases in Bulgaria, having been held since 2010. The event traditionally brings together all stakeholders, including medical specialists, patients, medical students, health authorities, and industry representatives, to present and discuss advancements in the diagnosis, treatment, and management of rare diseases, as well as access to innovations.
This year, the event will mark the 20th anniversary of the establishment of the Institute for Rare Diseases. The institute’s team has successfully contributed to the development of the national policy and strategy for rare diseases, providing information and consultations for patients and medical professionals. Additionally, they have successfully implemented various training programs, projects, and seminars.
Location: Hotel Imperial Plovdiv, Plovdiv
Early registration deadline: May 1, 2024
Late registration deadline: September 1, 2024
Abstract submission deadline: August 1, 2024
For contact with the organizing committee: congress@raredis.org
Additional information can be found at the XV National Conference on Rare Diseases and Orphan Drugs – Virtual Congress Center (raredis.org).
![]() Georgi Iskrov, Ralitsa Raycheva, Kostadin Kostadinov, Georgi Stefanov, Elena Mitova, Rumen Stefanov, and Stefan Stefanov, as part of the Institute for Rare Diseases and the Department of Social Medicine and Public Health at the Medical University – Plovdiv, actively participate in the Screen4Care project, which started in 2021. Screen4Care offers an innovative research approach to accelerate rare disease diagnosis, which is based on two central pillars: genetic newborn screening and digital technologies.
Georgi Iskrov, Ralitsa Raycheva, Kostadin Kostadinov, Georgi Stefanov, Elena Mitova, Rumen Stefanov, and Stefan Stefanov, as part of the Institute for Rare Diseases and the Department of Social Medicine and Public Health at the Medical University – Plovdiv, actively participate in the Screen4Care project, which started in 2021. Screen4Care offers an innovative research approach to accelerate rare disease diagnosis, which is based on two central pillars: genetic newborn screening and digital technologies.
Current research study “Are European Reference Networks for Rare Diseases Ready to Embrace Machine Learning?” has been executed by the Institute for Rare Diseases team together with Sandra Gillner and Carl Rudolf Blankart, KPM Center for Public Management, University of Bern, Swiss Institute for Translational and Entrepreneurial Medicine (Sitem-Insel), Switzerland; Edith Sky Gross and Gulcin Gumus, EURORDIS and is published in the prestigious international journal “Orphanet Journal of Rare Diseases” (Impact factor: 3.7)
The study involved 423 medical and other specialists associated with one of the 24 European Reference Networks. Participants express limited knowledge and experience with machine learning technologies. Surveyed individuals emphasize improved diagnostic accuracy as the most important potential benefit. Respondents support machine learning as recommended part of the rare diseases diagnostic process. Despite the presumed lack of experience, there is significant enthusiasm for the implementation of machine learning technologies, especially diagnostic tools targeting rare diseases. The study emphasizes the importance of early collaboration between healthcare professionals, software developers, industry, health authorities, and patient associations. This collaborative approach is considered crucial for building trust, improving efficiency, and increasing the acceptance of machine learning. Read the full article here.
Screen4Care project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking (JU) under Grant Agreement No. 101034427. The JU receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA.

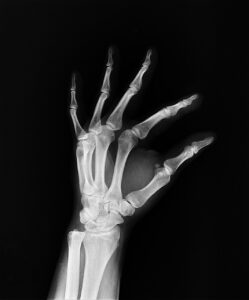 This study aims to describe a large cohort of Italian patients affected by osteogenesis imperfecta, providing a picture of the clinical bony and non-bony features and the molecular background to improve knowledge of the disease to inform appropriate management in clinical practice.
This study aims to describe a large cohort of Italian patients affected by osteogenesis imperfecta, providing a picture of the clinical bony and non-bony features and the molecular background to improve knowledge of the disease to inform appropriate management in clinical practice.
A total of 568 patients affected by osteogenesis imperfecta who received outpatient care at Istituto Ortopedico Rizzoli from 2006 to 2021 were considered in the present study.
Half of the patient population showed one or more deformities, and most of the patients had suffered a relatively low number of fractures (<10). An alteration in the sclera color was identified in 447 patients. Similarly, several extraskeletal features, like deafness, dental abnormalities, and cardiac problems, were investigated. Additionally, inheritance and genetic background were evaluated, showing that most of the patients have a positive family history.
This study supports the definition of a clear picture of the heterogeneous clinical manifestations leading to variable severity in terms of skeletal and extra-skeletal traits and of the genetic background of an Italian population of osteogenesis imperfecta patients. In this perspective, this clearly highlights the crucial role of standardized and structured collection of high-quality data in disease registries particularly in rare disease scenarios, helping clinicians in disease monitoring and follow-up to improve clinical practice. Read the full article here.
The image used in this article is sourced from: Pixabay
 In European Union countries, a disease affecting less than 5 people in 10,000 is considered rare. Due to the scarcity of expertise and the complexity of rare diseases, patients may go undiagnosed for years. Efforts are made to incorporate ORPHA nomenclature into health information systems, allowing coding for rare diseases. Previously, this nomenclature only covered specific rare diseases, leaving those with undiagnosed conditions without codes. Recognition of rare diseases status is crucial for patients, aiding in care reimbursement, providing social and psychological empowerment, and granting access to scientific advances, even without a precise diagnosis.
In European Union countries, a disease affecting less than 5 people in 10,000 is considered rare. Due to the scarcity of expertise and the complexity of rare diseases, patients may go undiagnosed for years. Efforts are made to incorporate ORPHA nomenclature into health information systems, allowing coding for rare diseases. Previously, this nomenclature only covered specific rare diseases, leaving those with undiagnosed conditions without codes. Recognition of rare diseases status is crucial for patients, aiding in care reimbursement, providing social and psychological empowerment, and granting access to scientific advances, even without a precise diagnosis.
The RD-CODE project aimed to identify undiagnosed patients in Health Information Systems for the purpose of gathering crucial epidemiological data. Undiagnosed patients were defined as those for whom no clinically-known disorder could be confirmed by an expert center despite all efforts to obtain a diagnosis. A multi-stakeholder panel of experts produced three recommendations for coding undiagnosed rare disease rare disease patients: 1/ Capture the diagnostic ascertainment for all rare disease cases; 2/ Use the newly created ORPHAcode (ORPHA:616874 “Rare disorder without a determined diagnosis after full investigation”), available in the Orphanet nomenclature: as the code is new, guidelines are essential to ensure its correct and homogeneous use for undiagnosed patients; 3/ Use additional descriptors in registries.
The recommendations can now be implemented in HIS (electronic health records and/or registries) and could be a game-changer for patients, clinicians and researchers in the field, enabling assessment of the rare disease population, including undiagnosed patients, adaptation of policy measures including financing for care and research programs, and to improved access of undiagnosed patients to research programs. Read the full article here.
 This study includes over 11.6M newborns screened (NBS) for Pompe Disease (PD) from 29 distinct universal screening programs across 8 countries and 4 continents. The birth prevalence of Pompe Disease is 1:18,711, with no evidence of difference across populations of European, Latin American, or Asian ancestry, though differences may exist for Pompe Disease subtypes. This study also compares these results, based on direct detection of disease and analyzed using a binomial method along with power analysis, with other methods for estimating the ‘frequency’ of rare genetic diseases.
This study includes over 11.6M newborns screened (NBS) for Pompe Disease (PD) from 29 distinct universal screening programs across 8 countries and 4 continents. The birth prevalence of Pompe Disease is 1:18,711, with no evidence of difference across populations of European, Latin American, or Asian ancestry, though differences may exist for Pompe Disease subtypes. This study also compares these results, based on direct detection of disease and analyzed using a binomial method along with power analysis, with other methods for estimating the ‘frequency’ of rare genetic diseases.
The primary objective of this study is to establish a new figure for prevalence at birth for Pompe disease by collecting and analyzing the largest relevant dataset to date. The other objective is to compare these results to previous analyses to offer a framework for evaluating ‘frequency’ data that can be applied to other rare, genetic diseases, along with methods to assess quality of estimates. Read the full article here.