
 Автозомно-доминантната левкодистрофия (ADLD) е изключително рядко, бавно прогресиращо невродегенеративно заболяване, свързано със загуба на бяло вещество в централната нервна система (ЦНС). Няколко години след първото му клинично описание е установено, че ADLD се причинява от варианти в гена LMNB1, които предизвикват неговата свръхекспресия в мозъка на пациентите. LMNB1 кодира Ламин B1, протеин от ядрената ламина. Ламин B1 регулира много клетъчни процеси като репликация на ДНК, организация на хроматина и стареене. Функциите му обаче все още не са напълно изяснени. Независимо от това, Ламин B1 заедно с другите ламини, които съставляват ядрената ламина, има ключова роля за поддържане на ядрената структура. Тъй като ядрото е динамична система, подложена както на биохимична, така и на механична регулация, е възможно промените в неговата структурна хомеостаза да се превърнат във функционални промени. В тази светлина този преглед има за цел да опише частит от доказателствата, получени до момента относно ефектите от свръхекспресията на LMNB1 върху клетъчната морфология и функционалност. Освен това, по-нататъшното изследване на ADLD морфо-функционалните последици е от съществено значение за по-доброто разбиране на това сложно заболяване. Прочетете цялата статия тук.
Автозомно-доминантната левкодистрофия (ADLD) е изключително рядко, бавно прогресиращо невродегенеративно заболяване, свързано със загуба на бяло вещество в централната нервна система (ЦНС). Няколко години след първото му клинично описание е установено, че ADLD се причинява от варианти в гена LMNB1, които предизвикват неговата свръхекспресия в мозъка на пациентите. LMNB1 кодира Ламин B1, протеин от ядрената ламина. Ламин B1 регулира много клетъчни процеси като репликация на ДНК, организация на хроматина и стареене. Функциите му обаче все още не са напълно изяснени. Независимо от това, Ламин B1 заедно с другите ламини, които съставляват ядрената ламина, има ключова роля за поддържане на ядрената структура. Тъй като ядрото е динамична система, подложена както на биохимична, така и на механична регулация, е възможно промените в неговата структурна хомеостаза да се превърнат във функционални промени. В тази светлина този преглед има за цел да опише частит от доказателствата, получени до момента относно ефектите от свръхекспресията на LMNB1 върху клетъчната морфология и функционалност. Освен това, по-нататъшното изследване на ADLD морфо-функционалните последици е от съществено значение за по-доброто разбиране на това сложно заболяване. Прочетете цялата статия тук.
 Диагностиката на редки болести и откриването на патогенни гени са революционизирани чрез секвениране на цял екзом и геном, но идентифицирането на причинителя(ите) от милионите във всеки индивид остава предизвикателство. Използването на фенотипизиране на пациенти и референтни познания за генотип-фенотип, заедно с различни данни като честота на алелите, сегрегация и прогнозирана патогенност, се оказа ефективна стратегия за справяне с този проблем. В тази статия са прегледани многобройните инструменти, които са разработени за автоматизиране на този подход и демонстриране на силата на такъв подход при няколко хиляди диагностицирани случая от проекта 100,000 Genomes. Обсъждат се още предизвикателствата, които трябва да бъдат преодолени, за да се подобрят нивата на откриване и да се помогне на повечето пациенти, които все още остават без молекулярна диагноза след най-съвременна геномна интерпретация. Прочетете цялата статия тук.
Диагностиката на редки болести и откриването на патогенни гени са революционизирани чрез секвениране на цял екзом и геном, но идентифицирането на причинителя(ите) от милионите във всеки индивид остава предизвикателство. Използването на фенотипизиране на пациенти и референтни познания за генотип-фенотип, заедно с различни данни като честота на алелите, сегрегация и прогнозирана патогенност, се оказа ефективна стратегия за справяне с този проблем. В тази статия са прегледани многобройните инструменти, които са разработени за автоматизиране на този подход и демонстриране на силата на такъв подход при няколко хиляди диагностицирани случая от проекта 100,000 Genomes. Обсъждат се още предизвикателствата, които трябва да бъдат преодолени, за да се подобрят нивата на откриване и да се помогне на повечето пациенти, които все още остават без молекулярна диагноза след най-съвременна геномна интерпретация. Прочетете цялата статия тук.
 Според данните на GLOBOCAN 2018 колоректалният рак представлява третото по честота онкологично заболяване в света. През 2018 г. заболеваемостта от рак на дебелото черво в световен мащаб е около 2 милиона, а броят на загиналите е близо 1 милион души. Голямата заболеваемост, в комбинация с високата смъртност и късното диагностициране на колоректалния карцином определят неговата социална значимост.
Според данните на GLOBOCAN 2018 колоректалният рак представлява третото по честота онкологично заболяване в света. През 2018 г. заболеваемостта от рак на дебелото черво в световен мащаб е около 2 милиона, а броят на загиналите е близо 1 милион души. Голямата заболеваемост, в комбинация с високата смъртност и късното диагностициране на колоректалния карцином определят неговата социална значимост.
Най-честите симптоми включват желязодефицитен тип анемия, промяна в дефекационния ритъм, редукция на тегло и прояви на чревна непроходимост. Не винаги обаче първата изява на клинична сиптоматика е от страна на дебелото черво, а и от други органи извън храносмилателната система. В тези случай ракът на дебелото черво обикновено е нелечим, вследствие развитие на екстраколонна дисеминация и развитие на съответна органна недостатъчност. Генетични фактори, заседналият начин на живот, затлъстяването, консумацията на червено месо, алкохол, тютюнопушене, някои метаболитни заболявания се считат за главни рискови фактори за възникването му.
Представяме случай на 47-годишна жена с хистологично верифицирани едновременни аденокарцином и карциноид на сигмоидното дебело черво, както и на фона на безсимптомно протичаща гастроезофагеална рефлуксна болест, усложнена с преканцерозата на хранопровода и стомаха – Баретов хранопровод. Прочетете цялата статия тук.
 Необходимостта от предоставяне на палиативни грижи за пациентите и техните семейства е от ключово значение за облекчаването на болките и страданията им, както и за съхраняване на възможността за достойно приключване на земният им път. Липсата на фокус от страна на законодателя за приемането на една цялостна нормативна рамка, която да включва в себе си формите, метода, начина на предоставяне и финансиране на палиативното лечение и грижа за нелечимо болните пациенти, създава усещането за отсъствие на държавата и провокира пациентите и техните семейства сами да търсят подкрепа и помощ за облекчаване на болките и страданията.
Необходимостта от предоставяне на палиативни грижи за пациентите и техните семейства е от ключово значение за облекчаването на болките и страданията им, както и за съхраняване на възможността за достойно приключване на земният им път. Липсата на фокус от страна на законодателя за приемането на една цялостна нормативна рамка, която да включва в себе си формите, метода, начина на предоставяне и финансиране на палиативното лечение и грижа за нелечимо болните пациенти, създава усещането за отсъствие на държавата и провокира пациентите и техните семейства сами да търсят подкрепа и помощ за облекчаване на болките и страданията.
Целта на настоящия анализ е да се представи съществуващата нормативната рамка, въз основа на която се предоставя палиативно лечение и грижа в Република България. Да се обобщи изчерпателността на нормативната дефиниция, както и да се направят релевантни препоръки за оптимизация на уредбата, с оглед констатираните празноти в нея. Прочетете цялата статия тук.
 Злокачествени заболявания като събирателна група от нозологични единици представляват значим социален и здравен проблем. Въпреки това, разглеждането на всички видове рак в една категория при изграждането на здравни политики е поставено под критика. Поради клиничните и епидемиологичните си характеристики, пациентите с редки тумори срещат в по-голяма степен бариери в достъпа си до иновативни медикаменти, съчетано с липса на експертиза в диагностично-лечебния процес. Изграждането на самостоятелни политики за тези пациенти изисква точна дефиниция на понятието.
Злокачествени заболявания като събирателна група от нозологични единици представляват значим социален и здравен проблем. Въпреки това, разглеждането на всички видове рак в една категория при изграждането на здравни политики е поставено под критика. Поради клиничните и епидемиологичните си характеристики, пациентите с редки тумори срещат в по-голяма степен бариери в достъпа си до иновативни медикаменти, съчетано с липса на експертиза в диагностично-лечебния процес. Изграждането на самостоятелни политики за тези пациенти изисква точна дефиниция на понятието.
Настоящият обзор цели да представи хронологичното развитие при дефинирането на понятието за редки тумори в контекста на изграждането на здравни политики в тази област. За целите на обзора е осъществено търсене по ключови думи „rare cancers”, „rare tumors” и „rare neoplasm” в базите данни PubMed/MEDLINE и ScienceDirect и Google Scholar. Откритите статии за организирани в три основни контекста на използване на термина „редки тумори“ – клинично базирани дефиниции; дефиниции, базирани на епидемиологични критерии и дефиниции, използвани в детската онкология. Прочетете цялата статия тук.
Д-р Костадин Димитров и Marie-Cecille Gaillard – проектен мениджър за наменклатурата на Orphanet и научен координатор на OD4RD, като част от проекта Orhanet Data for Rare Disease 2 (OD4RD2), който стартира през януари 2022г., представиха целите на проекта и резултатите от проучването си за предизвикателства и очаквания на български експертни центрове на XIV Национална конференция за редки болести и лекарства сираци.
Marie-Cecille Gaillard, представи организацията Orphanet, ползите от въвеждане на Orphanet кодирането и целите на проекта OD4RD 2.

Целите на проекта са: разработването на стандартизирани, оперативно съвместими данни, за диагностициране на редки болести чрез поддържане на номенклатурата на Orphanet и предоставяне на подкрепа за нейното прилагане в лечебни заведения, хармонизиране на събирането на данни в различни среди (здравни досиета, регистри) и държави чрез разпространение на най-добрите практики за кодиране и подпомагане вземането на решения основани на доказателства чрез предоставяне на изчерпателни данни относно редките болести.
Д-р Костадин Димитров от Национален център Orphanet България, Институт по редки болести представи резултатите от проучването сред български експертни центрове проведено през 2023г.
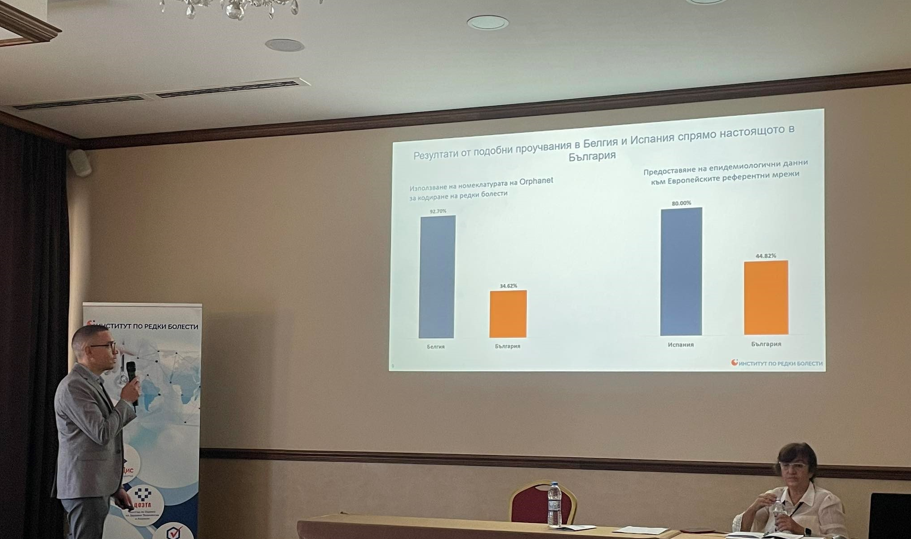
Цел на проучването е установяване на практиките, свързани с кодирането на редки болести в България към момента, както и анализ на предизвикателствата, които имат представителите на експертните центрове за редки болести при този процес за подобряване практическото използване на номенклатурата на Orphanet.
Резултатите от проучването ще послужат за установяване на предизвикателствата и потребностите пред партньорите и ще са основа за работата на създадения национален център ORPHANET България, чиято цел е да се ускори въвеждането и използването на ORPHA кодирането, в Експертните центрове за редки болести у нас. / с линк към страницата на OD4RD България/.
 Миелопролиферативните неоплазии (МПН) са хетерогенна група клонални заболявания на хемопоетичните стволови клетки, характеризиращи се с повишена пролиферация на клетки от миелоидния ред в костния мозък (КM). Ph-негативните MПН са най-честият рисков фактор за тромбоза на вена лиеналис (ТВЛ), включваща синдром на Бъд-Киари (БКС) и тромбоза на порталната вена (ТПВ). Редица проучвания разглеждат ролята на различните етиологични и рискови фактори за ТВЛ, включващи придобити протромботични състояния, наследствена тромбофилия и окални заболявания. Настоящото съобщение описва рядко срещан клиничен случай на пациентка с 3 рискови фактора за БКС: Ph-нег. МПН, причинена от придобита мутация JAK2V617F, наследствено нарушение на кръвосъсирването, фактор V Leiden мутация, и употреба на хормонална контрацепция.
Миелопролиферативните неоплазии (МПН) са хетерогенна група клонални заболявания на хемопоетичните стволови клетки, характеризиращи се с повишена пролиферация на клетки от миелоидния ред в костния мозък (КM). Ph-негативните MПН са най-честият рисков фактор за тромбоза на вена лиеналис (ТВЛ), включваща синдром на Бъд-Киари (БКС) и тромбоза на порталната вена (ТПВ). Редица проучвания разглеждат ролята на различните етиологични и рискови фактори за ТВЛ, включващи придобити протромботични състояния, наследствена тромбофилия и окални заболявания. Настоящото съобщение описва рядко срещан клиничен случай на пациентка с 3 рискови фактора за БКС: Ph-нег. МПН, причинена от придобита мутация JAK2V617F, наследствено нарушение на кръвосъсирването, фактор V Leiden мутация, и употреба на хормонална контрацепция.
Диагностично-терапевтичният алгоритъм при конкретната пациентка прецизно следва актуалните препоръки за поведение при това състояние, включващо и оценка на всички възможни протромботични рискови фактори.
Пациентката продължава своето регулярно проследяване от гастроентеролог и хематолог, поддържайки добро качество на живот без нови тромботични инциденти и нормални хематологични показатели.
Представеният клиничен случай потвърждава ролята на рутинния скрининг за МПН при ТВЛ. Откроява необходимост от извършване на скрининг за вродена тромбофилия, независимо от JAK2 статуса на пациента, поради вероятността за мултифакторна генеза на тромбообразуване при тези състояния, които от своя страна налагат прилагането на различни по механизъм на действие антитромботични терапии. Прочетете цялата статия тук.
Доц. Георги Искров, доц. Ралица Райчева и проф. Ричард Рьотгер като част от проекта Screen4Care, който стартира през 2021г., представиха първи резулти от проучванията си по време на XIV Национална конференция за редки болести и лекарства сираци – 30.09.2023. Основната цел на Screen4Care е да ускори поставянето на точна диагноза при хората с редки заболявания чрез прилагане на генетичен скрининг при новородени и разработване на иновативни дигитални решения.
Проф. Ричард Рьотгер е ръководител катедра по „Математика и компютърни науки“ на университета на южна Дания. Проф. Рьотгер се фокусира основно върху разработването на ефективни алгоритми за анализ на биологични мрежи и широкомащабни набори от биомедицински данни. Акцентът на неговата работа е върху неконтролираното машинно обучение и интегрирането на разнородни набори от данни. Проф. Рьотгер представи предизвикателствата и възможностите на прилагането на машинното самообучение в клиничната практика.

Георги Искров е доцент по икономика на здравеопазването в Медицински университет – Пловдив. От 2008 г. е част от екипа на Институт по редки болести, където участва в редица национални и европейски проекти. Доц. Искров представи резултатите от проучване проведено сред медицински специалисти работещи в европейските референтни мрежи относно готовността им за интегриране на машинно самообучен изкуствен интелект в практиката си.

Доц. Ралица Райчева е Магистър по макроикономика, доктор по медицина, научна специалност „Икономика на здравеопазването“. Доц. Райчева презентира резултати от изследване, публикувано в престижното международно списание: Frontiers in Public Health (Impact factor: 5,2; Cite score: 3,8) секция Public Health Policy. Проучването се фокусира върху предизвикателствата при картографирането на европейските бази данни за редки болести, свързани с технологии за скрининг, базирани на машинно самообучение (МС) и изкуствен интелект. Можете да прочетете цялата статията тук.
Проектът Screen4Care EU-IMI е финансиран от Innovative Medicines Initiative 2 Joint Undertaking (JU) съгласно споразумение за безвъзмездна подкрепа № 101034427. JU получава подкрепа от програмата за научни изследвания и иновации Horizon 2020 на Европейския съюз и EFPIA.

 Хемофилията е вродено нарушение в кръвосъсирването, което се дължи на дефицит или дефектни функции на коагулационен фактор VIII (FVIII) – наречено хемофилия А (ХА), или коагулационен фактор IX (FIX) – хемофилия B (ХB). Дефицитът на тези фактори е резултат от мутации в гените на FVIII или FIX. ХА се среща много по-често от ХB, като представлява приблизително 80-85% от случаите на всички пациенти с хемофилия. Средната честота е 24.6/100 000 мъже за ХА и 5/100 000 мъже при ХB.
Хемофилията е вродено нарушение в кръвосъсирването, което се дължи на дефицит или дефектни функции на коагулационен фактор VIII (FVIII) – наречено хемофилия А (ХА), или коагулационен фактор IX (FIX) – хемофилия B (ХB). Дефицитът на тези фактори е резултат от мутации в гените на FVIII или FIX. ХА се среща много по-често от ХB, като представлява приблизително 80-85% от случаите на всички пациенти с хемофилия. Средната честота е 24.6/100 000 мъже за ХА и 5/100 000 мъже при ХB.
ХА и ХB традиционно се разглеждат като клинично неразличими заболявания. Има някои доказателства, че тежък дефицит на FIX може да протича клинично по-леко от същия по тежест дефицит на фактор FVIII. Няколко възможни обяснения могат да се предположат – генетични дефекти, асоцииране с протромботични нарушения, характеристика на факторите на кръвосъсирване и други.
В заключение, въпреки клиничната неразличимост на двете заболявания, то при ХB се наблюдават някои разлики във фенотипно отношение, които изискват допълнителни изследвания върху механизмите, които протичат. Прочетете цялата статия тук.
 Хемоглобинопатиите са уникални сред всички генетични заболявания, защото в повечето случаи практически здравите носители могат да се идентифицират чрез общодостъпни кръвни тестове – кръвна картина, осмотична резистентност, типове хемоглобин ( първа група тестове) и да не се налага молекулярен анализ. При наличието на аномални хемоглобинови фракции се прилагат функционални изследвания, чрез които с голяма степен на достоверност се определя видът на патологичния хемоглобин и неговото клинично значение, без да се използват скъпоструващи молекулярни методи. Потвърждаването на наличието на генни мутации с молекулярни техники е необходимо само в малък процент от случаите – при т.нар. „рискови двойки” с цел профилактика на таласемия майор, за пренатална или преимплантационна диагностика – когато и двамата родители са суспектни или с доказано носителство на таласемия, или клинично значим патологичен хемоглобин.
Хемоглобинопатиите са уникални сред всички генетични заболявания, защото в повечето случаи практически здравите носители могат да се идентифицират чрез общодостъпни кръвни тестове – кръвна картина, осмотична резистентност, типове хемоглобин ( първа група тестове) и да не се налага молекулярен анализ. При наличието на аномални хемоглобинови фракции се прилагат функционални изследвания, чрез които с голяма степен на достоверност се определя видът на патологичния хемоглобин и неговото клинично значение, без да се използват скъпоструващи молекулярни методи. Потвърждаването на наличието на генни мутации с молекулярни техники е необходимо само в малък процент от случаите – при т.нар. „рискови двойки” с цел профилактика на таласемия майор, за пренатална или преимплантационна диагностика – когато и двамата родители са суспектни или с доказано носителство на таласемия, или клинично значим патологичен хемоглобин.
Чрез съпоставка и интерпретация на резултати от подходящи кръвни изследвания, анамнеза на пациента и вземане предвид на допълнителни фактори, правилната диагностика на хемоглобинопатиите с немолекулярни методи е от решаващо значение за предотвратяване на раждането на деца с тежката диагноза. Прочетете цялата статия тук.