
 Autosomal dominant leukodystrophy (ADLD) is an ultra-rare, slowly progressive neurodegenerative disorder associated with the loss of white matter in the central nervous system (CNS). Several years after its first clinical description, ADLD was found to be caused by variants in the LMNB1 gene that cause its overexpression in at least the brain of patients. LMNB1 encodes for Lamin B1, a protein of the nuclear lamina. Lamin B1 regulates many cellular processes such as DNA replication, chromatin organization, and senescence. However, its functions are not fully characterized yet. Nevertheless, Lamin B1 together with the other lamins that constitute the nuclear lamina has firstly the key role of maintaining the nuclear structure. Being the nucleus a dynamic system subject to both biochemical and mechanical regulation, it is conceivable that changes to its structural homeostasis might translate into functional alterations. Under this light, this review aims at describing the pieces of evidence that to date have been obtained regarding the effects of LMNB1 overexpression on cellular morphology and functionality. Moreover, further investigation on ADLD morpho-functional consequences is essential to better understand this complex disease and, possibly, other neurological disorders affecting CNS myelination are suggested. Read the full article here.
Autosomal dominant leukodystrophy (ADLD) is an ultra-rare, slowly progressive neurodegenerative disorder associated with the loss of white matter in the central nervous system (CNS). Several years after its first clinical description, ADLD was found to be caused by variants in the LMNB1 gene that cause its overexpression in at least the brain of patients. LMNB1 encodes for Lamin B1, a protein of the nuclear lamina. Lamin B1 regulates many cellular processes such as DNA replication, chromatin organization, and senescence. However, its functions are not fully characterized yet. Nevertheless, Lamin B1 together with the other lamins that constitute the nuclear lamina has firstly the key role of maintaining the nuclear structure. Being the nucleus a dynamic system subject to both biochemical and mechanical regulation, it is conceivable that changes to its structural homeostasis might translate into functional alterations. Under this light, this review aims at describing the pieces of evidence that to date have been obtained regarding the effects of LMNB1 overexpression on cellular morphology and functionality. Moreover, further investigation on ADLD morpho-functional consequences is essential to better understand this complex disease and, possibly, other neurological disorders affecting CNS myelination are suggested. Read the full article here.
 Rare disease diagnostics and disease gene discovery have been revolutionized by whole-exome and genome sequencing but identifying the causative variant(s) from the millions in each individual remains challenging. The use of deep phenotyping of patients and reference genotype-phenotype knowledge, alongside variant data such as allele frequency, segregation, and predicted pathogenicity, has proved an effective strategy to tackle this issue. In this article the numerous tools that have been developed to automate this approach and demonstrate the power of such an approach on several thousand diagnosed cases from the 100,000 Genomes Project are reviewed. Finally, the challenges that need to be overcome to improve detection rates and help the majority of patients that still remain without a molecular diagnosis after state-of-the-art genomic interpretation are discussed. Read the full article here.
Rare disease diagnostics and disease gene discovery have been revolutionized by whole-exome and genome sequencing but identifying the causative variant(s) from the millions in each individual remains challenging. The use of deep phenotyping of patients and reference genotype-phenotype knowledge, alongside variant data such as allele frequency, segregation, and predicted pathogenicity, has proved an effective strategy to tackle this issue. In this article the numerous tools that have been developed to automate this approach and demonstrate the power of such an approach on several thousand diagnosed cases from the 100,000 Genomes Project are reviewed. Finally, the challenges that need to be overcome to improve detection rates and help the majority of patients that still remain without a molecular diagnosis after state-of-the-art genomic interpretation are discussed. Read the full article here.
 According to GLOBOCAN 2018, colorectal cancer is the third most common cancer in the world. In 2018, the incidence of colon cancer worldwide is about 2 million, and the number of deaths is close to 1 million. The great morbidity, in combination with the high mortality and its late diagnosis, determine its social importance.
According to GLOBOCAN 2018, colorectal cancer is the third most common cancer in the world. In 2018, the incidence of colon cancer worldwide is about 2 million, and the number of deaths is close to 1 million. The great morbidity, in combination with the high mortality and its late diagnosis, determine its social importance.
The most common symptoms include iron-deficiency type anemia, change in defecation rhythm, weight reduction, and manifestations of intestinal obstruction. However, the first manifestation of clinical symptoms is not always from the large intestine but also from other organs outside the digestive system. In these cases, colon cancer is usually incurable due to the development of extracolonic dissemination and corresponding organ failure. Genetic factors, sedentary lifestyle, obesity, consumption of red meat, alcohol, smoking, and some metabolic diseases are considered the main risk factors for its occurrence.
We present the case of a 47-year-old woman with histologically verified simultaneous adenocarcinoma and carcinoid of the sigmoid colon, as well as the background of asymptomatic gastroesophageal reflux disease complicated by precancer of the esophagus and stomach – Barrett’s esophagus. Read the full article.
 The need to provide palliative care for patients and their families is key to alleviating their pain and suffering as well as preserving the possibility of a dignified end to their earthly journey. The lack of focus on the part of the legislator for the adoption of a comprehensive regulatory framework, which includes the forms, methods, and ways of providing and financing palliative treatment and care for terminally ill patients, creates a feeling of absence of the state and provokes patients and their families themselves to seek support and help to alleviate pain and suffering.
The need to provide palliative care for patients and their families is key to alleviating their pain and suffering as well as preserving the possibility of a dignified end to their earthly journey. The lack of focus on the part of the legislator for the adoption of a comprehensive regulatory framework, which includes the forms, methods, and ways of providing and financing palliative treatment and care for terminally ill patients, creates a feeling of absence of the state and provokes patients and their families themselves to seek support and help to alleviate pain and suffering.
The purpose of the present analysis is to present the existing regulatory framework, based on which palliative treatment and care are provided in the Republic of Bulgaria. To summarize the comprehensiveness of the normative definition as well as make relevant recommendations for optimization of the regulation in view of the gaps found in it. Read the full article here.
 Cancer diseases, as a group of pathological entities, represent a significant social and health problem. However, the consideration of all cancers in a single category in health policymaking has been subject to criticism. Due to their clinical and epidemiological characteristics, patients with rare cancers face obstacles in their access to innovative medicines, combined with a lack of expertise in the diagnostic and treatment processes. The establishment of specific policies for these patients requires a precise definition of the concept.
Cancer diseases, as a group of pathological entities, represent a significant social and health problem. However, the consideration of all cancers in a single category in health policymaking has been subject to criticism. Due to their clinical and epidemiological characteristics, patients with rare cancers face obstacles in their access to innovative medicines, combined with a lack of expertise in the diagnostic and treatment processes. The establishment of specific policies for these patients requires a precise definition of the concept.
This article aims to provide a chronological overview of the definition of “rare cancer” in the context of health policy development. A narrative literature review based on the keywords “rare cancers”, “rare tumors”, and “rare neoplasms” was conducted in the PubMed/MEDLINE, ScienceDirect, and Google Scholar databases. The identified articles were organised into three main contextual categories: clinical-based definitions, epidemiological-based definitions, and definitions used in pediatric oncology. Read the full article here.
Dr. Kostadin Dimitrov and Marie-Cecille Gaillard – Project Manager for Orphanet Nomenclature and Scientific Coordinator of OD4RD, as part of the Orphanet Data for Rare Disease 2 (OD4RD2) project, which started in January 2022, presented the project objectives and results from their research on challenges and expectations of Bulgarian expert centers at the XIV National Conference on Rare Diseases and Orphan Drugs.
Marie-Cecille Gaillard, presented the Orphanet organization, the benefits of implementing Orphanet coding and the goals of the OD4RD 2 project. The video can be viewed here.

The objectives of the project are: the development of standardized, interoperable data for the diagnosis of rare diseases by maintaining the Orphanet nomenclature and providing active support for its implementation in medical facilities, harmonizing the collection of data in different environments (health records, registries) and countries by disseminating best coding practices and supporting evidence-based decision-making by providing comprehensive data on rare diseases.
Dr. Kostadin Dimitrov, National Orphanet hub, Bulgaria presented the results of the study conducted with Bulgarian rare diseases expert centers in 2023.
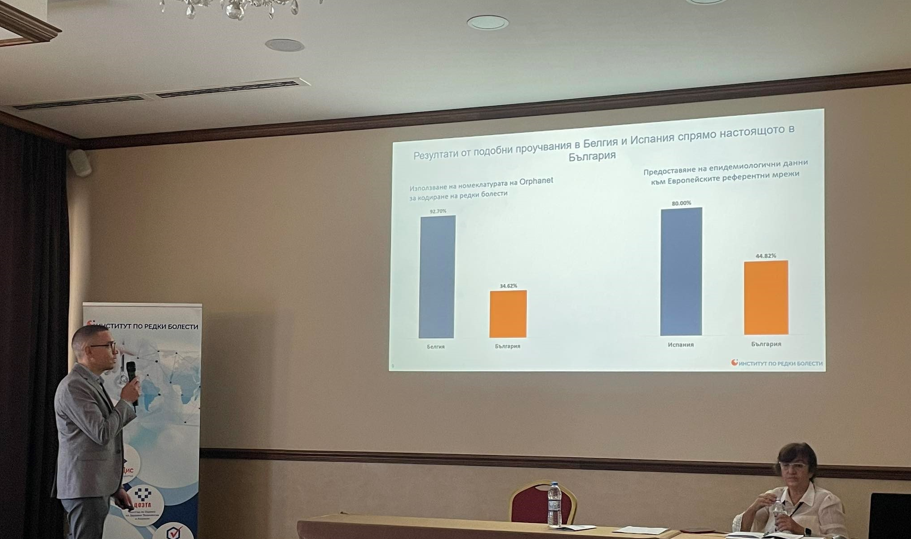
The objective of the study is to map current coding practices of rare diseases in Bulgaria, as well as to analyze the challenges that the expert centers representatives face in this process and reflect their proposals how to improve the use of Orphanet nomenclature.
The survey results will serve to identify the challenges and needs of our partners and will form a solid base for the work of the newly established National ORPHANET hub Bulgaria, whose goal is to accelerate the implementation and use of ORPHA coding in the expert centers for rare diseases in Bulgaria. / с линк към страницата
 Myeloproliferative neoplasias (MPNs) are a heterogeneous group of clonal diseases of hematopoietic stem cells characterized by increased proliferation of cells of the myeloid lineage in the bone marrow (BM). Ph-negative MPNs are the most common risk factor for vein thrombosis (VLT), including Budd-Chiari syndrome (BCS) and portal vein thrombosis (PVT). A number of studies have addressed the role of all etiologic and risk factors for VLT, including acquired prothrombotic conditions, hereditary thrombophilia, and local disease. The present communication describes a rare clinical case of a female patient with 3 risk factors for BCS: Ph-neg. MPN caused by acquired JAK2V617F mutation, inherited coagulation disorder, factor V Leiden mutation, and use of hormonal contraception.
Myeloproliferative neoplasias (MPNs) are a heterogeneous group of clonal diseases of hematopoietic stem cells characterized by increased proliferation of cells of the myeloid lineage in the bone marrow (BM). Ph-negative MPNs are the most common risk factor for vein thrombosis (VLT), including Budd-Chiari syndrome (BCS) and portal vein thrombosis (PVT). A number of studies have addressed the role of all etiologic and risk factors for VLT, including acquired prothrombotic conditions, hereditary thrombophilia, and local disease. The present communication describes a rare clinical case of a female patient with 3 risk factors for BCS: Ph-neg. MPN caused by acquired JAK2V617F mutation, inherited coagulation disorder, factor V Leiden mutation, and use of hormonal contraception.
The diagnostic-therapeutic algorithm for the specific patient exactly follows the current opinions on behavior in this condition, including the evaluation of all possible prothrombotic risk factors.
The patient continues his regular follow-up by a gastroenterologist and a hematologist, maintaining a good quality of life with no new thrombotic events and normal hematological parameters.
The presented clinical case confirms the role of routine screening for MPN in TVL. Screening for congenital thrombophilia is warranted regardless of the patient’s JAK2 status because of the likelihood of a multifactorial genesis of thrombosis in this condition, which on in turn necessitates the application of a different mechanism of antithrombotic action. Read the full article here.
Assoc. Prof. Georgi Iskrov, Assoc. Prof. Ralitsa Raicheva and Prof. Richard Rötger, as part of the Screen4Care project, which started in 2021, presented the first results of their studies during the XIV National Conference on Rare Diseases and Orphan Dugs – 30.09.2023. The main goal of Screen4Care project is to accelerate accurate diagnosis of people with rare diseases by implementing genetic screening in newborns and developing innovative digital solutions.
Prof. Richard Rötger is head of the Department of Mathematics and Computer Science at the University of Southern Denmark. Prof. Rötger mainly focuses on the development of efficient algorithms for the analysis of biological networks and large-scale biomedical datasets. His work focuses on unsupervised machine learning and integration of heterogeneous data sets. Prof. Rötger presented the challenges and opportunities for applying machine learning in clinical practice.

Georgi Iskrov is associate professor of healthcare economics at Medical University – Plovdiv. Since 2008, he has been part of the team of the Institute for Rare Diseases, where he participates in a number of national and European projects. Prof. Iskrov presented the results of a survey conducted among medical specialists working in European reference networks regarding their readiness to integrate machine-learned artificial intelligence in their practice.

Assoc. Prof. Raycheva has Master’s degree in Macroeconomics, PhD, scientific specialty “Healthcare Economics”. Associate Professor Raycheva presented the results of a study published in the international journal: Frontiers in Public Health (Impact factor: 5.2; Cite score: 3.8) Public Health Policy section. It focuses on the challenges when mapping European rare disease databases for screening technologies based on machine learning (ML) and artificial intelligence. You can read the published article here.
The Screen4Care EU-IMI project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement No 101034427. The JU receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA.

 Hemophilia is a congenital bleeding disorder characterized by a deficiency or functional defects of coagulation factor VIII (FVIII), called hemophilia A, or factor IX (FIX), called hemophilia B. The factor deficiencies are the result of mutations in the FVIII or FIX clotting factor genes. Hemophilia A is much more common than hemophilia B – estimated 80-85% of all hemophilia cases. Estimated prevalence at birth is 24.6/100 000 males for hemophilia A and 5/100 000 males for hemophilia B.
Hemophilia is a congenital bleeding disorder characterized by a deficiency or functional defects of coagulation factor VIII (FVIII), called hemophilia A, or factor IX (FIX), called hemophilia B. The factor deficiencies are the result of mutations in the FVIII or FIX clotting factor genes. Hemophilia A is much more common than hemophilia B – estimated 80-85% of all hemophilia cases. Estimated prevalence at birth is 24.6/100 000 males for hemophilia A and 5/100 000 males for hemophilia B.
Traditionally, hemophilia A and B have been considered clinically indistinguishable. There is some evidence that severe FIX deficiency may be clinically milder than the corresponding degree of FVIII deficiency. Several possible explanations for differences in disease severity can be hypothesized – genetic defects, associated prothrombotic abnormalities, clotting factor characteristics, etc.
In conclusion, despite the clinical indistinguishability of the two diseases, some phenotypic differences are observed in hemophilia B, which require further investigation of the underlying mechanisms. Read the full article here.
 Hemoglobinopathies are unique amongst all genetic diseases because the identification of healthy carriers is possible by commonly available blood tests – blood count, osmotic fragility, hemoglobin profile (first line tests) and molecular analysis is not required. If any abnormal hemoglobin fraction appears, functional tests are applied to determine with a pretty good degree of reliability the type of pathological hemoglobin and its clinical significance, avoiding the usage of expensive molecular methods. Confirmation of gene mutations presence with molecular techniques is only necessary in small number of cases – in so-called “couples at risk” for the purpose of prevention of thalassemia major, for prenatal or pre-implantation diagnosis – when both parents are thalassemia carriers (or suspected carriers), or carriers of clinically significant hemoglobin variant.
Hemoglobinopathies are unique amongst all genetic diseases because the identification of healthy carriers is possible by commonly available blood tests – blood count, osmotic fragility, hemoglobin profile (first line tests) and molecular analysis is not required. If any abnormal hemoglobin fraction appears, functional tests are applied to determine with a pretty good degree of reliability the type of pathological hemoglobin and its clinical significance, avoiding the usage of expensive molecular methods. Confirmation of gene mutations presence with molecular techniques is only necessary in small number of cases – in so-called “couples at risk” for the purpose of prevention of thalassemia major, for prenatal or pre-implantation diagnosis – when both parents are thalassemia carriers (or suspected carriers), or carriers of clinically significant hemoglobin variant.
By comparison and evaluation of the results of relevant blood tests, the patient’s history, and considering additional factors, the proper diagnosis of hemoglobinopathies with non-molecular methods is crucial for the prevention of a childbirth with a severe diagnosis. Read the full article here.