
 Antibody-dependent cellular cytotoxicity (ADCC) is the primary mechanism of actions for several marketed therapeutic antibodies (mAbs) and for many more in clinical trials. The ADCC efficacy is highly dependent on the ability of therapeutic mAbs to recruit effector cells such as natural killer cells, which induce the apoptosis of targeted cells. The recruitment of effector cells by mAbs is negatively affected by fucose modification of N-Glycans on the Fc; thus, utilization of afucosylated mAbs has been a trend for enhanced ADCC therapeutics. Most of afucosylated mAbs in clinical or commercial manufacturing were produced from Fut8-/- Chinese hamster ovary cells (CHO) host cells, generally generating low yields compared to wildtype CHO host. This study details the generation and characterization of two engineered CHOZN® cell lines, in which the enzyme involved in guanosine diphosphate (GDP)-fucose synthesis, GDP mannose-4,6-dehydratase (Gmds) and GDP-L-fucose synthase (FX), was knocked out. The top host cell lines for each of the knockouts, FX-/- and Gmds-/-, were selected based on growth robustness, bulk MSX selection tolerance, production titer, fucosylation level, and cell stability. We tested the production of two proprietary IgG1 mAbs in the engineered host cells, and found that the titers were comparable to CHOZN® cells. The mAbs generated from either KO cell line exhibited loss of fucose modification, leading to significantly boosted FcγRIIIa binding and ADCC effects. Our data demonstrated that both FX-/- and Gmds-/- host cells could replace Fut8-/- CHO cells for clinical manufacturing of antibody therapeutics. For more information click here.
Antibody-dependent cellular cytotoxicity (ADCC) is the primary mechanism of actions for several marketed therapeutic antibodies (mAbs) and for many more in clinical trials. The ADCC efficacy is highly dependent on the ability of therapeutic mAbs to recruit effector cells such as natural killer cells, which induce the apoptosis of targeted cells. The recruitment of effector cells by mAbs is negatively affected by fucose modification of N-Glycans on the Fc; thus, utilization of afucosylated mAbs has been a trend for enhanced ADCC therapeutics. Most of afucosylated mAbs in clinical or commercial manufacturing were produced from Fut8-/- Chinese hamster ovary cells (CHO) host cells, generally generating low yields compared to wildtype CHO host. This study details the generation and characterization of two engineered CHOZN® cell lines, in which the enzyme involved in guanosine diphosphate (GDP)-fucose synthesis, GDP mannose-4,6-dehydratase (Gmds) and GDP-L-fucose synthase (FX), was knocked out. The top host cell lines for each of the knockouts, FX-/- and Gmds-/-, were selected based on growth robustness, bulk MSX selection tolerance, production titer, fucosylation level, and cell stability. We tested the production of two proprietary IgG1 mAbs in the engineered host cells, and found that the titers were comparable to CHOZN® cells. The mAbs generated from either KO cell line exhibited loss of fucose modification, leading to significantly boosted FcγRIIIa binding and ADCC effects. Our data demonstrated that both FX-/- and Gmds-/- host cells could replace Fut8-/- CHO cells for clinical manufacturing of antibody therapeutics. For more information click here.
 Multiple sulfatase deficiency (MSD) is an ultra-rare neurodegenerative disorder caused by pathogenic variants in SUMF1. This gene encodes formylglycine-generating enzyme (FGE), a protein required for sulfatase activation. The clinical course of MSD results from additive effect of each sulfatase deficiency, including metachromatic leukodystrophy (MLD), several mucopolysaccharidoses (MPS II, IIIA, IIID, IIIE, IVA, VI), chondrodysplasia punctata, and X-linked ichthyosis. While it is known that affected individuals demonstrate a complex and severe phenotype, the genotype-phenotype relationship and detailed clinical course is unknown. We report on 35 cases enrolled in our retrospective natural history study. Neurologic function was longitudinally assessed with retrospective scales. Biochemical and computational modeling of novel SUMF1 variants was performed. Genotypes were classified based on predicted functional change, and each individual was assigned a genotype severity score. The median age at symptom onset was 0.25 years; median age at diagnosis was 2.7 years; and median age at death was 13 years. All individuals demonstrated developmental delay, and only a subset of individuals attained ambulation and verbal communication. All subjects experienced an accumulating systemic symptom burden. Earlier age at symptom onset and severe variant pathogenicity correlated with poor neurologic outcomes. Using retrospective deep phenotyping and detailed variant analysis, we defined the natural history of MSD. We found that attenuated cases can be distinguished from severe cases by age of onset, attainment of ambulation, and genotype. Results from this study can help inform prognosis and facilitate future study design. For more information click here.
Multiple sulfatase deficiency (MSD) is an ultra-rare neurodegenerative disorder caused by pathogenic variants in SUMF1. This gene encodes formylglycine-generating enzyme (FGE), a protein required for sulfatase activation. The clinical course of MSD results from additive effect of each sulfatase deficiency, including metachromatic leukodystrophy (MLD), several mucopolysaccharidoses (MPS II, IIIA, IIID, IIIE, IVA, VI), chondrodysplasia punctata, and X-linked ichthyosis. While it is known that affected individuals demonstrate a complex and severe phenotype, the genotype-phenotype relationship and detailed clinical course is unknown. We report on 35 cases enrolled in our retrospective natural history study. Neurologic function was longitudinally assessed with retrospective scales. Biochemical and computational modeling of novel SUMF1 variants was performed. Genotypes were classified based on predicted functional change, and each individual was assigned a genotype severity score. The median age at symptom onset was 0.25 years; median age at diagnosis was 2.7 years; and median age at death was 13 years. All individuals demonstrated developmental delay, and only a subset of individuals attained ambulation and verbal communication. All subjects experienced an accumulating systemic symptom burden. Earlier age at symptom onset and severe variant pathogenicity correlated with poor neurologic outcomes. Using retrospective deep phenotyping and detailed variant analysis, we defined the natural history of MSD. We found that attenuated cases can be distinguished from severe cases by age of onset, attainment of ambulation, and genotype. Results from this study can help inform prognosis and facilitate future study design. For more information click here.
 Interest in drug development for rare diseases has expanded dramatically since the Orphan Drug Act was passed in 1983, with 40% of new drug approvals in 2019 targeting orphan indications. However, limited quantitative understanding of natural history and disease progression hinders progress and increases the risks associated with rare disease drug development. Use of international data standards can assist in data harmonization and enable data exchange, integration into larger datasets, and a quantitative understanding of disease natural history. The US Food and Drug Administration (FDA) requires the use of Clinical Data Interchange Consortium (CDISC) Standards in new drug submissions to help the agency efficiently and effectively receive, process, review, and archive submissions, as well as to help integrate data to answer research questions. Such databases have been at the core of biomarker qualification efforts and fit-for-purpose models endorsed by the regulators. We describe the development of CDISC therapeutic area user guides for Duchenne muscular dystrophy and Huntington’s disease through Critical Path Institute consortia. These guides describe formalized data structures and controlled terminology to map and integrate data from different sources. This will result in increased standardization of data collection and allow integration and comparison of data from multiple studies. Integration of multiple data sets enables a quantitative understanding of disease progression, which can help overcome common challenges in clinical trial design in these and other rare diseases. Ultimately, clinical data standardization will lead to a faster path to regulatory approval of urgently needed new therapies for patients. For more information click here.
Interest in drug development for rare diseases has expanded dramatically since the Orphan Drug Act was passed in 1983, with 40% of new drug approvals in 2019 targeting orphan indications. However, limited quantitative understanding of natural history and disease progression hinders progress and increases the risks associated with rare disease drug development. Use of international data standards can assist in data harmonization and enable data exchange, integration into larger datasets, and a quantitative understanding of disease natural history. The US Food and Drug Administration (FDA) requires the use of Clinical Data Interchange Consortium (CDISC) Standards in new drug submissions to help the agency efficiently and effectively receive, process, review, and archive submissions, as well as to help integrate data to answer research questions. Such databases have been at the core of biomarker qualification efforts and fit-for-purpose models endorsed by the regulators. We describe the development of CDISC therapeutic area user guides for Duchenne muscular dystrophy and Huntington’s disease through Critical Path Institute consortia. These guides describe formalized data structures and controlled terminology to map and integrate data from different sources. This will result in increased standardization of data collection and allow integration and comparison of data from multiple studies. Integration of multiple data sets enables a quantitative understanding of disease progression, which can help overcome common challenges in clinical trial design in these and other rare diseases. Ultimately, clinical data standardization will lead to a faster path to regulatory approval of urgently needed new therapies for patients. For more information click here.
 Оn October 3 from 16:00 to 19:30 will be held the online event of World Alliance of Pituitary Organizations (WAPO), of which the Pituitary Association is a member. The event will be translated in Russian and English. Registration is free. For more information click here.
Оn October 3 from 16:00 to 19:30 will be held the online event of World Alliance of Pituitary Organizations (WAPO), of which the Pituitary Association is a member. The event will be translated in Russian and English. Registration is free. For more information click here.
 Our goal is to contribute both improving the care and life of people with rare diseases, and the quality of our work. We invite all participants in the XI National Conference for Rare Diseases and Orphan Drugs to share their impressions by filling out a short survey. Its purpose is to comprehend the point of view and opinion about the conference. Thank you for your cooperation!
Our goal is to contribute both improving the care and life of people with rare diseases, and the quality of our work. We invite all participants in the XI National Conference for Rare Diseases and Orphan Drugs to share their impressions by filling out a short survey. Its purpose is to comprehend the point of view and opinion about the conference. Thank you for your cooperation!
 This report describes a new strategy for the care of patients with osteogenesis imperfecta, based on an interdisciplinary team working. Thereby, we aim at fulfilling three main goals: offering thorough coordinated management for all, and improving physical activity and quality of life of the patients. With rare diseases such as osteogenesis imperfecta (OI), patients and their family often suffer from inadequate recognition of their disease, poor care coordination and incomplete information. A coordinated interdisciplinary approach is one possible solution for providing both comprehensive and cost-effective care, with benefits for patient satisfaction. Poor physical activity and impaired quality of life represent a considerable burden for these patients. To better address these issues, in 2012 we created an interdisciplinary team for the management of OI patients in our University Hospital Centre (CHUV, Lausanne University Hospital,). In this article we describe the implementation of this interdisciplinary care strategy for patients suffering from OI, and its impact on their physical activity and quality of life. For more information click here.
This report describes a new strategy for the care of patients with osteogenesis imperfecta, based on an interdisciplinary team working. Thereby, we aim at fulfilling three main goals: offering thorough coordinated management for all, and improving physical activity and quality of life of the patients. With rare diseases such as osteogenesis imperfecta (OI), patients and their family often suffer from inadequate recognition of their disease, poor care coordination and incomplete information. A coordinated interdisciplinary approach is one possible solution for providing both comprehensive and cost-effective care, with benefits for patient satisfaction. Poor physical activity and impaired quality of life represent a considerable burden for these patients. To better address these issues, in 2012 we created an interdisciplinary team for the management of OI patients in our University Hospital Centre (CHUV, Lausanne University Hospital,). In this article we describe the implementation of this interdisciplinary care strategy for patients suffering from OI, and its impact on their physical activity and quality of life. For more information click here.
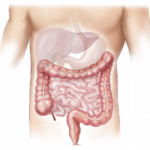 Short bowel syndrome (SBS) is a rare disease that results from extensive resection of the intestine. When the remaining absorption surface of the intestine cannot absorb enough macronutrients, micronutrients, and water, SBS results in intestinal failure (IF). Patients with SBS who suffer from IF require parenteral nutrition for survival, but long-term parenteral nutrition may lead to complications such as catheter sepsis and metabolic diseases. Spontaneous intestinal adaptation occurs weeks to months after resection, resulting in hyperplasia of the remnant gut, modification of gut hormone levels, dysbiosis, and hyperphagia. Oral nutrition and presence of the colon are two major positive drivers for this adaptation. This review aims to summarize the current knowledge of the mechanisms underlying spontaneous intestinal adaptation, particularly in response to modifications of luminal content, including nutrients. In the future, dietary manipulations could be used to treat SBS. For more information click here.
Short bowel syndrome (SBS) is a rare disease that results from extensive resection of the intestine. When the remaining absorption surface of the intestine cannot absorb enough macronutrients, micronutrients, and water, SBS results in intestinal failure (IF). Patients with SBS who suffer from IF require parenteral nutrition for survival, but long-term parenteral nutrition may lead to complications such as catheter sepsis and metabolic diseases. Spontaneous intestinal adaptation occurs weeks to months after resection, resulting in hyperplasia of the remnant gut, modification of gut hormone levels, dysbiosis, and hyperphagia. Oral nutrition and presence of the colon are two major positive drivers for this adaptation. This review aims to summarize the current knowledge of the mechanisms underlying spontaneous intestinal adaptation, particularly in response to modifications of luminal content, including nutrients. In the future, dietary manipulations could be used to treat SBS. For more information click here.
 Thalassemia is characterized by a defect in the synthesis of one or more of the globin subunits of hemoglobin. This defect results in imbalance in the α/β-globin chain ratio, ineffective erythropoiesis, chronic hemolytic anemia, and iron overload. With advances in diagnosis, treatment, and transfusion support, the prognosis of patients with thalassemia has improved over the past few decades. An increasing number of patients with thalassemia is living with long-term complications, including cardiomyopathy, chronic liver disease, endocrinopathy, and infections. In this paper, we review common complications that bring the patient with thalassemia to urgent or emergent medical attention. We also discuss the aspects of emergency care that are most relevant while caring for the patient with thalassemia in the emergency department. For more information click here.
Thalassemia is characterized by a defect in the synthesis of one or more of the globin subunits of hemoglobin. This defect results in imbalance in the α/β-globin chain ratio, ineffective erythropoiesis, chronic hemolytic anemia, and iron overload. With advances in diagnosis, treatment, and transfusion support, the prognosis of patients with thalassemia has improved over the past few decades. An increasing number of patients with thalassemia is living with long-term complications, including cardiomyopathy, chronic liver disease, endocrinopathy, and infections. In this paper, we review common complications that bring the patient with thalassemia to urgent or emergent medical attention. We also discuss the aspects of emergency care that are most relevant while caring for the patient with thalassemia in the emergency department. For more information click here.
 To coordinate care effectively for rare conditions, we need to understand what coordinated care means. This review aimed to define coordinated care and identify components of coordinated care within the context of rare diseases; by drawing on evidence from chronic conditions. A systematic scoping review. We included reviews that reported or defined and outlined components of coordinated care for chronic or rare conditions. Thematic analysis was used to develop a definition and identify components or care coordination. Stakeholder consultations (three focus groups with patients, carers and healthcare professionals with experience of rare conditions) were held to further explore the relevance of review findings for rare conditions. Coordinated care is multi-faceted and has both generic and context-specific components. Findings outline many ways in which care may be coordinated for both rare and common chronic conditions. Findings can help to develop and eventually test different ways of coordinating care for people with rare and common chronic conditions. For more information click here.
To coordinate care effectively for rare conditions, we need to understand what coordinated care means. This review aimed to define coordinated care and identify components of coordinated care within the context of rare diseases; by drawing on evidence from chronic conditions. A systematic scoping review. We included reviews that reported or defined and outlined components of coordinated care for chronic or rare conditions. Thematic analysis was used to develop a definition and identify components or care coordination. Stakeholder consultations (three focus groups with patients, carers and healthcare professionals with experience of rare conditions) were held to further explore the relevance of review findings for rare conditions. Coordinated care is multi-faceted and has both generic and context-specific components. Findings outline many ways in which care may be coordinated for both rare and common chronic conditions. Findings can help to develop and eventually test different ways of coordinating care for people with rare and common chronic conditions. For more information click here.
 Chronic myeloid leukemia (CML) is a rare disease in childhood. While hematopoietic stem cell transplant (HSCT) was the treatment of choice for CML prior to 2000, the introduction of tyrosine kinase inhibitors (TKIs) changed the management of this disease. This population-based analysis was conducted in the province of Ontario, Canada to gather information on treatment choices and outcomes of childhood CML. Using a provincial childhood cancer registry and retrospective review of patient medical records for patients < 18 years diagnosed with CML between 1985 and 2018, data on presenting features, treatment, and outcomes were collected from 52 patients. Given the increased mortality associated with HSCT in our cohort, further advances in HSCT may be required to outweigh the benefits of a TKI monotherapy approach in the majority of childhood CML patients. We believe HSCT should be considered in only a limited subset of pediatric patients with CML. For more information click here.
Chronic myeloid leukemia (CML) is a rare disease in childhood. While hematopoietic stem cell transplant (HSCT) was the treatment of choice for CML prior to 2000, the introduction of tyrosine kinase inhibitors (TKIs) changed the management of this disease. This population-based analysis was conducted in the province of Ontario, Canada to gather information on treatment choices and outcomes of childhood CML. Using a provincial childhood cancer registry and retrospective review of patient medical records for patients < 18 years diagnosed with CML between 1985 and 2018, data on presenting features, treatment, and outcomes were collected from 52 patients. Given the increased mortality associated with HSCT in our cohort, further advances in HSCT may be required to outweigh the benefits of a TKI monotherapy approach in the majority of childhood CML patients. We believe HSCT should be considered in only a limited subset of pediatric patients with CML. For more information click here.