
 European Reference Networks (ERNs) were created under the 2011 Directive on Patient Rights’ in Cross-Border Healthcare. They are based on the voluntary cooperation of Member States that contribute to the ERNs’ activities in accordance with their national legislation. ERNs are operational since March 2017. To ensure a proper and sustainable functioning of the ERNs and to reap all benefits for patients suffering from rare and low prevalence complex diseases across the EU, the ERNs need to be linked in a clear and stable way to the healthcare systems of the Member States. These are issues of key importance and demand tangible actions. This Statement aims to give incentives to Member States to further enhance the integration process based on the input provided by the Working Group on Integration. Member States are encouraged to facilitate the integration of ERNs to their healthcare systems by: assessing and if needed adapting or updating the national policy and/or legal framework; creating appropriate (clear and well-defined) patients’ pathways in order to improve the care and management of patients with complex or rare diseases; developing clear systems for referral to ERNs; developing a clear strategy for communicating and disseminating information about ERNs; reflecting on the means to best support. For more information click here.
European Reference Networks (ERNs) were created under the 2011 Directive on Patient Rights’ in Cross-Border Healthcare. They are based on the voluntary cooperation of Member States that contribute to the ERNs’ activities in accordance with their national legislation. ERNs are operational since March 2017. To ensure a proper and sustainable functioning of the ERNs and to reap all benefits for patients suffering from rare and low prevalence complex diseases across the EU, the ERNs need to be linked in a clear and stable way to the healthcare systems of the Member States. These are issues of key importance and demand tangible actions. This Statement aims to give incentives to Member States to further enhance the integration process based on the input provided by the Working Group on Integration. Member States are encouraged to facilitate the integration of ERNs to their healthcare systems by: assessing and if needed adapting or updating the national policy and/or legal framework; creating appropriate (clear and well-defined) patients’ pathways in order to improve the care and management of patients with complex or rare diseases; developing clear systems for referral to ERNs; developing a clear strategy for communicating and disseminating information about ERNs; reflecting on the means to best support. For more information click here.
 The EURORDIS Photo Award is one of the 14 Black Pearl Awards that recognise the stars of the rare disease community, as nominated by the rare disease community and members of the public. Whether you are an amateur photographer or took that one great photo that illustrates the challenges or joy of living with a rare disease, you can submit your photo by 16 January to be in with a chance of winning the EURORDIS Photo Award 2020! For more information click here.
The EURORDIS Photo Award is one of the 14 Black Pearl Awards that recognise the stars of the rare disease community, as nominated by the rare disease community and members of the public. Whether you are an amateur photographer or took that one great photo that illustrates the challenges or joy of living with a rare disease, you can submit your photo by 16 January to be in with a chance of winning the EURORDIS Photo Award 2020! For more information click here.
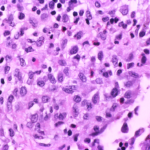 Langerhans cell histiocytosis (LCH) is a rare disease of unknown aetiology. While it may affect any organ of the body, few cases of solitary lung involvement are published in the literature. Here, we report a rare case of pulmonary LCH (PLCH) in an adult. A 52-year-old male presented to hospital in July 2018 with complaints of progressively worsening cough with sputum, breathlessness, easy fatigability, and loss of appetite since 2016, and a 32-year history of heavy cigarette smoking (average 30 cigarettes/d). Physical examination showed only weakened breathing sounds and wheezing during lung auscultation. Chest computed tomography (CT) showed irregular micronodules and multiple thin-walled small holes. Respiratory function tests showed a slight decrease. Ultrasonic cardiogram showed mild tricuspid regurgitation and no pulmonary hypertension. Fibreoptic bronchoscopy was performed with transbronchial biopsies from the basal segment of right lower lobe. LCH was confirmed by immunohistochemistry. The final diagnosis was PLCH without extra-pulmonary involvement. We suggested smoking cessation treatment. A 3-mo follow-up chest CT scan showed clear absorption of the nodule and thin-walled small holes. The symptoms of cough and phlegm had improved markedly and appetite had improved. There was no obvious dyspnoea. Imaging manifestations of nodules, cavitating nodules, and thick-walled or thin-walled cysts prompted suspicion of PLCH and lung biopsy for diagnosis. For more information click here.
Langerhans cell histiocytosis (LCH) is a rare disease of unknown aetiology. While it may affect any organ of the body, few cases of solitary lung involvement are published in the literature. Here, we report a rare case of pulmonary LCH (PLCH) in an adult. A 52-year-old male presented to hospital in July 2018 with complaints of progressively worsening cough with sputum, breathlessness, easy fatigability, and loss of appetite since 2016, and a 32-year history of heavy cigarette smoking (average 30 cigarettes/d). Physical examination showed only weakened breathing sounds and wheezing during lung auscultation. Chest computed tomography (CT) showed irregular micronodules and multiple thin-walled small holes. Respiratory function tests showed a slight decrease. Ultrasonic cardiogram showed mild tricuspid regurgitation and no pulmonary hypertension. Fibreoptic bronchoscopy was performed with transbronchial biopsies from the basal segment of right lower lobe. LCH was confirmed by immunohistochemistry. The final diagnosis was PLCH without extra-pulmonary involvement. We suggested smoking cessation treatment. A 3-mo follow-up chest CT scan showed clear absorption of the nodule and thin-walled small holes. The symptoms of cough and phlegm had improved markedly and appetite had improved. There was no obvious dyspnoea. Imaging manifestations of nodules, cavitating nodules, and thick-walled or thin-walled cysts prompted suspicion of PLCH and lung biopsy for diagnosis. For more information click here.
 Idiopathic pulmonary fibrosis (IPF) is a chronic pulmonary disease that is complicated by diagnostic challenges, multiple comorbidities, and a poor prognosis. Although considered a relatively rare disease, healthcare costs are substantial and disproportionate to the incidence and prevalence of the disease. The comorbidities associated with IPF not only complicate treatment strategies but also increase the burden for patients via higher costs and undesirable health outcomes. Historically, pharmacologic treatment options for IPF have been limited and are often associated with low efficacy. Two drugs approved for IPF, nintedanib and pirfenidone, offer promise for improving health outcomes and survival during the course of the disease. Considerations of cost and adverse events are important when planning treatment options. Optimizing care through patient-centered care management programs can improve outcomes and health-related quality of life for patients. Such programs emphasize communication between healthcare professionals and patients in order to educate patients on their condition, so they can make informed healthcare decisions. Disease registries can be important tools for optimizing data collection and analysis for a disease with limited incidence and prevalence. For more information click here.
Idiopathic pulmonary fibrosis (IPF) is a chronic pulmonary disease that is complicated by diagnostic challenges, multiple comorbidities, and a poor prognosis. Although considered a relatively rare disease, healthcare costs are substantial and disproportionate to the incidence and prevalence of the disease. The comorbidities associated with IPF not only complicate treatment strategies but also increase the burden for patients via higher costs and undesirable health outcomes. Historically, pharmacologic treatment options for IPF have been limited and are often associated with low efficacy. Two drugs approved for IPF, nintedanib and pirfenidone, offer promise for improving health outcomes and survival during the course of the disease. Considerations of cost and adverse events are important when planning treatment options. Optimizing care through patient-centered care management programs can improve outcomes and health-related quality of life for patients. Such programs emphasize communication between healthcare professionals and patients in order to educate patients on their condition, so they can make informed healthcare decisions. Disease registries can be important tools for optimizing data collection and analysis for a disease with limited incidence and prevalence. For more information click here.
 Rett syndrome (RTT) is an early-onset neurodevelopmental disorder that primarily affects females, resulting in severe cognitive and physical disabilities, and is one of the most prevalent causes of intellectual disability in females. More than fifty years after the first publication on Rett syndrome, and almost two decades since the first report linking RTT to the MECP2 gene, the research community’s effort is focused on obtaining a better understanding of the genetics and the complex biology of RTT and Rett-like phenotypes without MECP2 mutations. Herein, we review the current molecular genetic studies, which investigate the genetic causes of RTT or Rett-like phenotypes which overlap with other genetic disorders and document the swift evolution of the techniques and methodologies employed. This review also underlines the clinical and genetic heterogeneity of the Rett syndrome spectrum and provides an overview of the RTT-related genes described to date, many of which are involved in epigenetic gene regulation, neurotransmitter action or RNA transcription/translation. Finally, it discusses the importance of including both phenotypic and genetic diagnosis to provide proper genetic counselling from a patient’s perspective and the appropriate treatment. For more information click here.
Rett syndrome (RTT) is an early-onset neurodevelopmental disorder that primarily affects females, resulting in severe cognitive and physical disabilities, and is one of the most prevalent causes of intellectual disability in females. More than fifty years after the first publication on Rett syndrome, and almost two decades since the first report linking RTT to the MECP2 gene, the research community’s effort is focused on obtaining a better understanding of the genetics and the complex biology of RTT and Rett-like phenotypes without MECP2 mutations. Herein, we review the current molecular genetic studies, which investigate the genetic causes of RTT or Rett-like phenotypes which overlap with other genetic disorders and document the swift evolution of the techniques and methodologies employed. This review also underlines the clinical and genetic heterogeneity of the Rett syndrome spectrum and provides an overview of the RTT-related genes described to date, many of which are involved in epigenetic gene regulation, neurotransmitter action or RNA transcription/translation. Finally, it discusses the importance of including both phenotypic and genetic diagnosis to provide proper genetic counselling from a patient’s perspective and the appropriate treatment. For more information click here.
![]() Fabry disease and Pompe disease are rare lysosomal storage disorders that belong to a heterogeneous group of more than 200 distinct inborn metabolic diseases. Mutations followed by loss of function of enzymes or transporters that are localised in the acidic environment of the lysosome may result in degradation of many substrates, such as glycosaminoglycans, glycosphingolipids, glycogen, cholesterol, oligosaccharides, glycoproteins, and peptides, or the excretion of the products degraded by the lysosome. Our aim was to identify the oral signs and symptoms of Fabry disease and Pompe disease from a systematic review made using MEDLINE/PubMed, and a hand search for relevant articles, following the PRISMA guidelines. Both diseases show various craniofacial and oral changes, including supernumerary teeth, dental agenesis, angiokeratoma, and telangiectases in Fabry disease; and macroglossia, teeth fusion, and taurodontism in Pompe disease. Common clinical signs of Fabry disease include hyposalivation, hypohidrosis, and xerophthalmia, and a generally reduced physical resilience was apparent in patients with Pompe disease. Oral and craniofacial changes in patients with both diseases extend over their entire lifetime and can be detected even in an infant. Lysosomal storage diseases should be taken into consideration in the differential diagnosis of relevant diverse symptoms, because treatment, when available, is most effective when started early. The main therapeutic concepts are enzymatic replacement for Pompe disease, whereas patients with Fabry disease require additional oral chaperone treatment or enzyme replacement. For more information click here.
Fabry disease and Pompe disease are rare lysosomal storage disorders that belong to a heterogeneous group of more than 200 distinct inborn metabolic diseases. Mutations followed by loss of function of enzymes or transporters that are localised in the acidic environment of the lysosome may result in degradation of many substrates, such as glycosaminoglycans, glycosphingolipids, glycogen, cholesterol, oligosaccharides, glycoproteins, and peptides, or the excretion of the products degraded by the lysosome. Our aim was to identify the oral signs and symptoms of Fabry disease and Pompe disease from a systematic review made using MEDLINE/PubMed, and a hand search for relevant articles, following the PRISMA guidelines. Both diseases show various craniofacial and oral changes, including supernumerary teeth, dental agenesis, angiokeratoma, and telangiectases in Fabry disease; and macroglossia, teeth fusion, and taurodontism in Pompe disease. Common clinical signs of Fabry disease include hyposalivation, hypohidrosis, and xerophthalmia, and a generally reduced physical resilience was apparent in patients with Pompe disease. Oral and craniofacial changes in patients with both diseases extend over their entire lifetime and can be detected even in an infant. Lysosomal storage diseases should be taken into consideration in the differential diagnosis of relevant diverse symptoms, because treatment, when available, is most effective when started early. The main therapeutic concepts are enzymatic replacement for Pompe disease, whereas patients with Fabry disease require additional oral chaperone treatment or enzyme replacement. For more information click here.
 This guideline was designed to provide service providers and users with an evidence-based set of current best practice guidelines for people and their families and carers, living with epidermolysis bullosa (EB). A systematic literature review relating to the podiatric care of patients with EB was undertaken. Search terms were used, for which the most recent articles relating to podiatric treatment were identified from as early as 1979 to the present day, across seven electronic search engines: MEDLINE, Wiley Online Library, Google Scholar, Athens, ResearchGate, Net and PubFacts.com. The Scottish Intercollegiate Guidelines Network (SIGN) methodology was used. The first guideline draft was analysed and discussed by clinical experts, methodologists and patients and their representatives at four panel meetings. The resulting document went through an external review process by a panel of experts, other healthcare professionals, patient representatives and lay reviewers. The final document will be piloted in three different centres in the U.K. and Australia. Following an EB community international survey the outcomes indicated six main areas that the community indicated as a priority to foot management. These include blistering and wound management, exploring the most suitable footwear and hosiery for EB, management of dystrophic nails, hyperkeratosis (callus), maintaining mobility and fusion of toes (pseudosyndactyly). The evidence here is limited but several interventions currently practised by podiatrists show positive outcomes. For more information click here.
This guideline was designed to provide service providers and users with an evidence-based set of current best practice guidelines for people and their families and carers, living with epidermolysis bullosa (EB). A systematic literature review relating to the podiatric care of patients with EB was undertaken. Search terms were used, for which the most recent articles relating to podiatric treatment were identified from as early as 1979 to the present day, across seven electronic search engines: MEDLINE, Wiley Online Library, Google Scholar, Athens, ResearchGate, Net and PubFacts.com. The Scottish Intercollegiate Guidelines Network (SIGN) methodology was used. The first guideline draft was analysed and discussed by clinical experts, methodologists and patients and their representatives at four panel meetings. The resulting document went through an external review process by a panel of experts, other healthcare professionals, patient representatives and lay reviewers. The final document will be piloted in three different centres in the U.K. and Australia. Following an EB community international survey the outcomes indicated six main areas that the community indicated as a priority to foot management. These include blistering and wound management, exploring the most suitable footwear and hosiery for EB, management of dystrophic nails, hyperkeratosis (callus), maintaining mobility and fusion of toes (pseudosyndactyly). The evidence here is limited but several interventions currently practised by podiatrists show positive outcomes. For more information click here.
 Achondroplasia is the most common form of disproportionate short stature and might affect not only the quality of life of the affected child but also that of the parents. The aim is to investigate the quality of life of children with achondroplasia from child- and parent perspective as well as the parental quality of life. Forty-seven children with achondroplasia and 73 parents from a German patient organization participated. We assessed children’s quality of life using the generic Peds QL 4.0™ as self-reports for children aged 8-14 and parent-reports for children aged 4-14 years. Parental quality of life we assessed using the short-form 8-questionnaire. Achondroplasia is chronically debilitating. Thus special efforts are needed to address patients’ and parent’s quality of life needs. This special health condition may influence the daily life of the entire family because they have to adapt to the child’s particular needs. Therefore, clinicians should not only focus on the child’s quality of life but also those of the parents. For more information click here.
Achondroplasia is the most common form of disproportionate short stature and might affect not only the quality of life of the affected child but also that of the parents. The aim is to investigate the quality of life of children with achondroplasia from child- and parent perspective as well as the parental quality of life. Forty-seven children with achondroplasia and 73 parents from a German patient organization participated. We assessed children’s quality of life using the generic Peds QL 4.0™ as self-reports for children aged 8-14 and parent-reports for children aged 4-14 years. Parental quality of life we assessed using the short-form 8-questionnaire. Achondroplasia is chronically debilitating. Thus special efforts are needed to address patients’ and parent’s quality of life needs. This special health condition may influence the daily life of the entire family because they have to adapt to the child’s particular needs. Therefore, clinicians should not only focus on the child’s quality of life but also those of the parents. For more information click here.
 This year on 6-8 of December 2019 in Athens, Greece the Duchenne Data Foundation, with the support of the World Duchenne Organization, is launching the 3rd edition of the Duchenne Patient Academy. Over the past years, the Duchenne world has been changing due to a fast evolving research and regulatory landscape. For this, the need for adaptive and proactive Duchenne advocates has increased. In this intensive two-day training session, both Next Generation Young Advocates and Experienced Advocates will receive a parallel training and updates to set a strong base for current and future global advocacy.This Duchenne Masterclass will focus on setting the foundation of advocacy and gaining a deeper understanding of the Duchenne field in terms of regulatory, HTA, clinical trial, care, research and industry developments. For more information click here.
This year on 6-8 of December 2019 in Athens, Greece the Duchenne Data Foundation, with the support of the World Duchenne Organization, is launching the 3rd edition of the Duchenne Patient Academy. Over the past years, the Duchenne world has been changing due to a fast evolving research and regulatory landscape. For this, the need for adaptive and proactive Duchenne advocates has increased. In this intensive two-day training session, both Next Generation Young Advocates and Experienced Advocates will receive a parallel training and updates to set a strong base for current and future global advocacy.This Duchenne Masterclass will focus on setting the foundation of advocacy and gaining a deeper understanding of the Duchenne field in terms of regulatory, HTA, clinical trial, care, research and industry developments. For more information click here.
 NCATS is dedicated to engaging the patient community throughout the translational science process. The NCATS Toolkit for Patient-Focused Therapy Development was created to provide a collection of online resources that can help patient groups advance through the process of therapy development and provide them with the tools they need to advance medical research. Launched in September 2017, the Toolkit includes resources that have been developed primarily for the rare diseases community to facilitate therapeutics research and development. Since early 2016, NCATS has worked with a diverse group of partners in the rare diseases community to conduct an extensive landscape analysis of available tools. These resources were defined, characterized and organized in a centralized portal that can be helpful to all patient groups regardless of how far along in the research and development process they might be. For more information click here.
NCATS is dedicated to engaging the patient community throughout the translational science process. The NCATS Toolkit for Patient-Focused Therapy Development was created to provide a collection of online resources that can help patient groups advance through the process of therapy development and provide them with the tools they need to advance medical research. Launched in September 2017, the Toolkit includes resources that have been developed primarily for the rare diseases community to facilitate therapeutics research and development. Since early 2016, NCATS has worked with a diverse group of partners in the rare diseases community to conduct an extensive landscape analysis of available tools. These resources were defined, characterized and organized in a centralized portal that can be helpful to all patient groups regardless of how far along in the research and development process they might be. For more information click here.