
 Миастения гравис (MG) е рядко състояние, което уврежда функцията на нервно-мускулното съединение на скелетните мускули, наблюдавано по-рядко при деца. Причините включват автоимунна MG, вродени миастенични синдроми и преходна неонатална миастения гравис. Симптомите на слабост, хипотония и умора могат да се обяснят с по-чести причини, поради което децата със заболяването MG обикновено изпитват забавяне на лечението с тежки последици. Това води до прогресиране на заболяването и сериозни усложнения, включително миастенични кризи и екзацербации. В този доклад са описани 5 случая на MG, които илюстрират клинични и генетични предизвикателства при поставяне на диагноза и последици от забавенато ѝ. Прочетете цялата статия тук.
Миастения гравис (MG) е рядко състояние, което уврежда функцията на нервно-мускулното съединение на скелетните мускули, наблюдавано по-рядко при деца. Причините включват автоимунна MG, вродени миастенични синдроми и преходна неонатална миастения гравис. Симптомите на слабост, хипотония и умора могат да се обяснят с по-чести причини, поради което децата със заболяването MG обикновено изпитват забавяне на лечението с тежки последици. Това води до прогресиране на заболяването и сериозни усложнения, включително миастенични кризи и екзацербации. В този доклад са описани 5 случая на MG, които илюстрират клинични и генетични предизвикателства при поставяне на диагноза и последици от забавенато ѝ. Прочетете цялата статия тук.
 Генетиката допринася значително за предразположението към идиопатична белодробна фиброза (IPF). Генетичните изследвания при спорадични и фамилни заболявания са идентифицирали няколко варианта, свързани с IPF, главно в гени, свързани с теломерите и повърхностно активни протеини.
Генетиката допринася значително за предразположението към идиопатична белодробна фиброза (IPF). Генетичните изследвания при спорадични и фамилни заболявания са идентифицирали няколко варианта, свързани с IPF, главно в гени, свързани с теломерите и повърхностно активни протеини.
Последните проучвания включват гени, участващи в поддържането на теломерите, защита на гостоприемника, клетъчно-клетъчна адхезия. Както често срещаните, така и редките генетични варианти допринасят за общия риск от IPF; въпреки това, докато често срещаните варианти (т.е. полиморфизми) представляват по-голямата част от наследствеността на спорадичните заболявания, редките варианти (т.е. мутации), главно в гените, свързани с теломерите, са основните фактори, допринасящи за наследствеността на фамилните заболявания. Вероятно е генетичните фактори също да повлияят за протичането и прогнозата на заболяването. Последните данни показват, че IPF споделя генетични асоциации – и вероятно някои патогенетични механизми – с други фиброзни белодробни заболявания. Прочетете цялата статия тук.
 Емболите, причинени от сърдечни миксоми, се появяват предимно в кръвноносните съдове на сърцето или мозъчно-съдовата система и рядко в кръвоносните съдове на долните крайници. В този доклад се представя рядък случай на пациент с миксом на лявото предсърдие, чийто десен долен крайник страда от остра исхемия поради туморни фрагменти. В този случай се съобщава за 81-годишна жена с остра исхемия на десен долен крайник. Доплеровата ултразвукова ехография не показва сигнал за кръвен ток по дясната феморална артерия. Компютърна томографска ангиография показва оклузия на дясната обща феморална артерия. Трансторакалната ехокардиограма разкрива маса в лявото предсърдие. Извършена е емболектомия на бедрената артерия, последвана от торакотомия с резекция на тумора под обща анестезия на седмия следоперативен ден. Туморът е патологично потвърден като предсърден миксом. Прочетете цялата статия тук.
Емболите, причинени от сърдечни миксоми, се появяват предимно в кръвноносните съдове на сърцето или мозъчно-съдовата система и рядко в кръвоносните съдове на долните крайници. В този доклад се представя рядък случай на пациент с миксом на лявото предсърдие, чийто десен долен крайник страда от остра исхемия поради туморни фрагменти. В този случай се съобщава за 81-годишна жена с остра исхемия на десен долен крайник. Доплеровата ултразвукова ехография не показва сигнал за кръвен ток по дясната феморална артерия. Компютърна томографска ангиография показва оклузия на дясната обща феморална артерия. Трансторакалната ехокардиограма разкрива маса в лявото предсърдие. Извършена е емболектомия на бедрената артерия, последвана от торакотомия с резекция на тумора под обща анестезия на седмия следоперативен ден. Туморът е патологично потвърден като предсърден миксом. Прочетете цялата статия тук.
 Атипичният хемолитично – уремичен синдром (aHUS) е рядко заболяване, с оскъдни съобщения за неврологични прояви. Исхемични кортикални инфаркти, съпътстващи представянето на aHUS, не са описани при възрастни пациенти.
Атипичният хемолитично – уремичен синдром (aHUS) е рядко заболяване, с оскъдни съобщения за неврологични прояви. Исхемични кортикални инфаркти, съпътстващи представянето на aHUS, не са описани при възрастни пациенти.
В този доклад се представя 46-годишен мъж с остро влошаващ се психичен статус и прогресивна слабост на фона на дългогодишна хипертония и известна аортна дисекация тип В. Спешното образно изследване показва двустранни мултифокални мултитериториални исхемични инфаркти, свързани с емболичен източник или хиперкоагулационно състояние. Общите изследвания показват микроангиопатична хемолитична анемия и остро бъбречно увреждане. Започва се плазмафереза при предполагаема тромботична тромбоцитопенична пурпура. Започва се лечение с инхибитор на комплемента и пациентът постепенно се възстановява. Генетичното изследване потвърждава патогенна мутация, CFHR1 хомозиготна делеция.
Острите мултифокални мултитериториални исхемични инфаркти и системната тромботична микроангиопатия могат да бъдат проява на aHUS и със свързана генетична мутация, дори при възрастни пациенти. Прочетете цялата статия тук.
 Дефицитът на декарбоксилаза на ароматна l-аминокиселина (AADC) е рядко автозомно рецесивно неврометаболитно разстройство, причинено от двуалелни патогенни варианти в DDC гена и характеризиращо се главно със забавяне на развитието, хипотония и окулогирични кризи. Ранната диагностика е от решаващо значение за правилното лечение на пациента; въпреки това много пациенти остават погрешно диагностицирани или недиагностицирани поради рядкостта и клиничната хетерогенност на разстройството, особено при по-леките форми.
Дефицитът на декарбоксилаза на ароматна l-аминокиселина (AADC) е рядко автозомно рецесивно неврометаболитно разстройство, причинено от двуалелни патогенни варианти в DDC гена и характеризиращо се главно със забавяне на развитието, хипотония и окулогирични кризи. Ранната диагностика е от решаващо значение за правилното лечение на пациента; въпреки това много пациенти остават погрешно диагностицирани или недиагностицирани поради рядкостта и клиничната хетерогенност на разстройството, особено при по-леките форми.
В това проучване се прилага подход за секвениране на екзоми чрез скрининг на 2000 педиатрични пациенти с нарушения на неврологичното развитие, за да се идентифицират възможни нови варианти на AADC и пациенти с дефицит на AADC. Идентифицирани са пет различни варианта на DDC при два несвързани индивида. Вариантите са класифицирани като доброкачествени от клас I и следователно не са причинители съгласно насоките на ACMG/AMP. Пациентите в това проучване, носещи варианти на DDC, представят клинични прояви, които не се припокриват точно с класическите симптоми, проявявани от най-тежките случаи на дефицит на AADC. Въпреки това, скрининговите данни, получени от секвениране на екзоми при пациенти, характеризиращи се с широк спектър от симптоми, свързани с нарушения на неврологичното развитие, могат да помогнат за идентифициране на пациенти с дефицит на AADC, особено когато се прилагат при по-големи кохорти. Прочетете цялата статия тук.
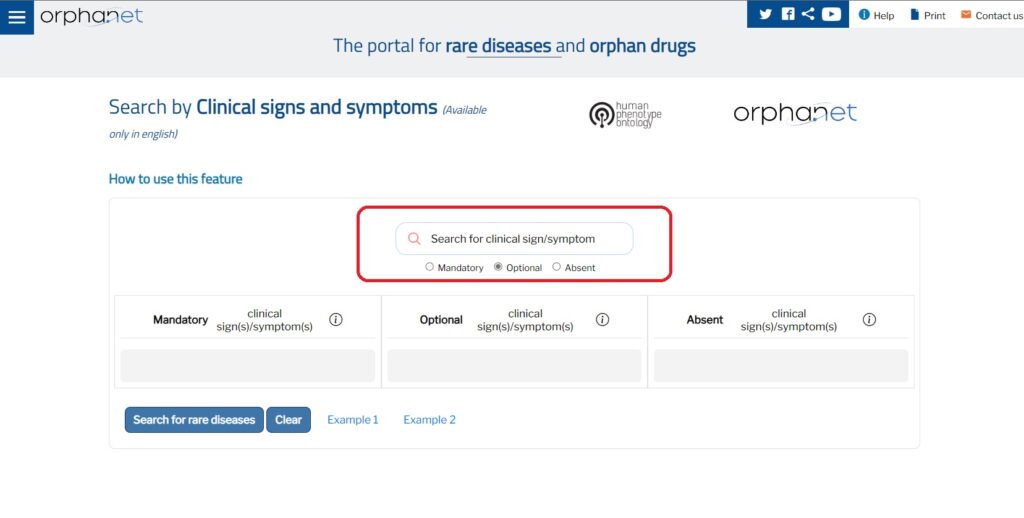
 В портала за редки болести и лекарства сираци на Orphanet (https://www.orpha.net/consor/cgi-bin/index.php) вече можете да ползвате ново приложение на английски език за търсене на рядко заболяване по клинични признаци или симптоми.
В портала за редки болести и лекарства сираци на Orphanet (https://www.orpha.net/consor/cgi-bin/index.php) вече можете да ползвате ново приложение на английски език за търсене на рядко заболяване по клинични признаци или симптоми.
Orphanet предоставя клинично описание на редки заболявания, използвайки набор от клинични признаци и симптоми. Това описание, базирано на случаи, публикувани в биомедицинската литература, използва фенотипните аномалии, посочени в онтологията на човешкия фенотип (Human Phenotype Ontology, HPO).
Приложението е достъпно на следния линк: https://clinicalsigns.orphanet.app като в раздела „Клинични признаци и симптоми“ можете да извършвате търсенето чрез въвеждане на признаци и симптоми, които Ви интересуват. В лентата за търсене въведете клиничния признак/симптом, който искате да търсите, и изберете съответния термин на HPO.
Предоставената информация е изчислена за цялата популация пациенти в рутинната медицинска практика.
 В Съединените щати рядко заболяване се определя като състояние, което засяга по-малко от 200 000 души. Като цяло редките болести засягат около 30 милиона американци. Значителна част от редките заболявания се причиняват от генетичен дефект; това обаче може да остане недиагностицирано. За да обслужва по-добре тези пациенти, програмата на Mayo Clinic за редки и недиагностицирани заболявания е създадена под егидата на Центъра за индивидуализирана медицина, целяща да интегрира геномиката в практиката, включително целеви генетични тестове, изследвания и образование.
В Съединените щати рядко заболяване се определя като състояние, което засяга по-малко от 200 000 души. Като цяло редките болести засягат около 30 милиона американци. Значителна част от редките заболявания се причиняват от генетичен дефект; това обаче може да остане недиагностицирано. За да обслужва по-добре тези пациенти, програмата на Mayo Clinic за редки и недиагностицирани заболявания е създадена под егидата на Центъра за индивидуализирана медицина, целяща да интегрира геномиката в практиката, включително целеви генетични тестове, изследвания и образование.
Изпълнението на Програмата за редки и недиагностицирани заболявания започва през 2018 г., а звеното за генетично изследване и консултиране стартира през 2020 г. в подкрепа на разширяването на програмата. В момента 29 клинични индикации за редки заболявания са включени в 11 специализирани отделения.
Изпълнението на програмата за редки и недиагностицирани болести дава възможност на субспециализираните практики да усъвършенстват експертния си опит в областта на редките болести, където генетичните консултанти не са били внедрени в практиката. Достъпа до генетично изследване и консултиране може да разшири диагностицирането на пациентите с редки заболявания, което води до по-добрa задравна помощ, както и разширяване на възможностите за научни изследвания. Прочетете цялата статия тук.
 Болестта на Rosai-Dorfman е рядко доброкачествено хистиоцитно заболяване, характеризиращо се в повечето случаи с безболезнена цервикална аденопатия. По-малко от 10% от екстранодалните случаи включват костни лезии. Първичната костна болест на Rosai-Dorfman при липса на нодално засягане е изключително рядка.
Болестта на Rosai-Dorfman е рядко доброкачествено хистиоцитно заболяване, характеризиращо се в повечето случаи с безболезнена цервикална аденопатия. По-малко от 10% от екстранодалните случаи включват костни лезии. Първичната костна болест на Rosai-Dorfman при липса на нодално засягане е изключително рядка.
В този доклад е представен случай на 48-годишен мъж с прогресираща дясностранна оталгия, шум в ушите, световъртеж и загуба на слуха. Литична лезия на дясната темпорална кост се открива при образна диагностика. Резекцията на лезията и хистопатологичното изследване разкриват болест на Rosai-Dorfman.
Първичните костни лезии при болестта на Rosai-Dorfman са нетипична проява на рядко заболяване. Това е вторият докладван случай на болест на Rosai-Dorfman, възникваща в темпоралната кост. Този казус разкрива, че болестта на Rosai-Dorfman трябва да се има предвид при пациенти с възпалителни/литични лезии на темпоралната кост, в случаите, когато са изключени инфекция и злокачествено заболяване. Прочетете цялата статия тук.
 Синдромът на White Sutton е рядко автозомно доминантно заболяване, което е резултат от de novo мутация. Фенотипът се характеризира с широк спектър от когнитивни дисфункции и изоставане в развитието. Загубата на слуха често се споменава като един от симптомите на това рядко заболяване, но подробностите обикновено са оскъдни. В този доклад е показан случай на дете от мъжки пол, засегнато от синдрома на White Sutton и сензоневрална загуба на слуха. Към днешна дата настоящият случай е първото описание на загуба на слуха, дължаща се на слуховата невропатия при синдрома на White Sutton. Следователно цялостната оценка на слуха е задължителна при всички пациенти със синдром на White Sutton, за да се разпознае възможна слухова невропатия и след това да се избегне погрешна диагноза или погрешно клинично лечение. Прочетете цялата статия тук.
Синдромът на White Sutton е рядко автозомно доминантно заболяване, което е резултат от de novo мутация. Фенотипът се характеризира с широк спектър от когнитивни дисфункции и изоставане в развитието. Загубата на слуха често се споменава като един от симптомите на това рядко заболяване, но подробностите обикновено са оскъдни. В този доклад е показан случай на дете от мъжки пол, засегнато от синдрома на White Sutton и сензоневрална загуба на слуха. Към днешна дата настоящият случай е първото описание на загуба на слуха, дължаща се на слуховата невропатия при синдрома на White Sutton. Следователно цялостната оценка на слуха е задължителна при всички пациенти със синдром на White Sutton, за да се разпознае възможна слухова невропатия и след това да се избегне погрешна диагноза или погрешно клинично лечение. Прочетете цялата статия тук.
 Хемиплегичната мигрена е рядко заболяване и данните за педиатричната популация са още по-редки, тъй като често се екстраполират от смесени кохорти.
Хемиплегичната мигрена е рядко заболяване и данните за педиатричната популация са още по-редки, тъй като често се екстраполират от смесени кохорти.
Целта на това проучване е да опише кохорта от педиатрични пациенти с генетично потвърдена фамилна хемиплегична мигрена (FHM). Познаването на корелациите генотип-фенотип може да предложи прогностични фактори, свързани с тежки фенотипове.
Избрани са пациенти, които отговарят на Международната класификация на главоболието, които са имали молекулярна диагноза на FHM и чийто първи пристъп е настъпил на възраст под 18 години.
Данните от проучването показват, че повечето от пациентите с ранно начало на FHM изпитват редки и леки пристъпи, които се подобряват с времето. Освен това, клиничният курс не разкрива нито появата на нови неврологични разстройства, нито влошаване на основното неврологични или когнитивни функции. Прочетете цялата статия тук.