
 Myasthenia gravis (MG) is a rare condition that impairs function at the neuromuscular junction of skeletal muscles, seen less commonly in children. Causes include autoimmune MG, congenital myasthenic syndromes, and transient neonatal myasthenia gravis. Symptoms of weakness, hypotonia, and fatigability can be reasonably explained by more common causes, thus children with MG disorders commonly experience delays in treatment with severe consequences. This leads to the progression of disease and serious complications including myasthenic crises and exacerbations. In this report are 5 cases of MG, which illustrate clinical and genetic challenges in establishing diagnosis and subsequent consequences of delayed diagnosis described. Read the full article here.
Myasthenia gravis (MG) is a rare condition that impairs function at the neuromuscular junction of skeletal muscles, seen less commonly in children. Causes include autoimmune MG, congenital myasthenic syndromes, and transient neonatal myasthenia gravis. Symptoms of weakness, hypotonia, and fatigability can be reasonably explained by more common causes, thus children with MG disorders commonly experience delays in treatment with severe consequences. This leads to the progression of disease and serious complications including myasthenic crises and exacerbations. In this report are 5 cases of MG, which illustrate clinical and genetic challenges in establishing diagnosis and subsequent consequences of delayed diagnosis described. Read the full article here.
 Genetics contributes substantially to the susceptibility to idiopathic pulmonary fibrosis (IPF). Genetic studies in sporadic and familial disease have identified several IPF-associated variants, mainly in telomere-related and surfactant protein genes.
Genetics contributes substantially to the susceptibility to idiopathic pulmonary fibrosis (IPF). Genetic studies in sporadic and familial disease have identified several IPF-associated variants, mainly in telomere-related and surfactant protein genes.
Recent studies implicate genes involved in telomere maintenance, host defence, cell growth, cell-cell adhesion. Both common and rare genetic variants contribute to the overall risk of IPF; however, while common variants (i.e. polymorphisms) account for most of the heritability of sporadic disease, rare variants (i.e. mutations), mainly in telomere-related genes, are the main contributors to the heritability of familial disease. Genetic factors are likely to also influence disease behaviour and prognosis. Finally, recent data suggest that IPF shares genetic associations – and probably some pathogenetic mechanisms – with other fibrotic lung diseases. Read the full article here.
 Emboli caused by cardiac myxomas mostly occur in the cardiovascular or cerebrovascular systems and rarely in the lower extremity vasculature. In this report a rare case of a patient with left atrial myxoma (LAM) whose right lower extremity (RLE) suffered from acute ischemia due to tumor fragments is introduced. In this case a 81-year-old female presented with acute ischemia of RLE is reported. Color Doppler ultrasound showes no blood flow signal far from the RLE femoral artery. Computed tomography angiography shows an occlusion of the right common femoral artery. A transthoracic echocardiogram reveals a left atrial mass. Femoral artery embolectomy is performed, followed by thoracotomy with tumor resection under general anesthesia on postoperative day seven. The tumor is pathologically confirmed as an atrial myxoma. Read the full article here.
Emboli caused by cardiac myxomas mostly occur in the cardiovascular or cerebrovascular systems and rarely in the lower extremity vasculature. In this report a rare case of a patient with left atrial myxoma (LAM) whose right lower extremity (RLE) suffered from acute ischemia due to tumor fragments is introduced. In this case a 81-year-old female presented with acute ischemia of RLE is reported. Color Doppler ultrasound showes no blood flow signal far from the RLE femoral artery. Computed tomography angiography shows an occlusion of the right common femoral artery. A transthoracic echocardiogram reveals a left atrial mass. Femoral artery embolectomy is performed, followed by thoracotomy with tumor resection under general anesthesia on postoperative day seven. The tumor is pathologically confirmed as an atrial myxoma. Read the full article here.
 Atypical hemolytic uremic syndrome (aHUS) is a rare disease, with scarce reports of neurologic manifestations in the acute setting. Ischemic cortical infarcts concurrently with aHUS presentation have not been described in adult patients.
Atypical hemolytic uremic syndrome (aHUS) is a rare disease, with scarce reports of neurologic manifestations in the acute setting. Ischemic cortical infarcts concurrently with aHUS presentation have not been described in adult patients.
A 46-year-old male presents with acutely declining mental status and progressive weakness, in the setting of longstanding hypertension and known type B aortic dissection. Urgent neuroimaging shows bilateral multifocal multiterritorial ischemic infarcts, concerning for an embolic source or hypercoagulable state. Systemic workup is notable for microangiopathic hemolytic anemia and acute kidney injury. Empiric plasmapheresis is initiated for presumed thrombotic thrombocytopenic purpura. Treatment with complement inhibitor is started and patient gradually recovers. Genetic testing confirms a pertinent pathogenic mutation, CFHR1 homozygous deletion.
Acute multifocal multiterritorial ischemic infarcts and systemic thrombotic microangiopathy may be a manifestation of aHUS, and with associated genetic mutation, even in adult population. Read the full article here.
 Aromatic l-amino acid decarboxylase (AADC) deficiency is a rare autosomal recessive neurometabolic disorder caused by biallelic pathogenic variants in the DDC gene and mainly characterized by developmental delay, hypotonia, and oculogyric crises. Early diagnosis is crucial for correct patient management; however, many patients remain misdiagnosed or undiagnosed due to the rarity and clinical heterogeneity of the disorder especially in the milder forms.
Aromatic l-amino acid decarboxylase (AADC) deficiency is a rare autosomal recessive neurometabolic disorder caused by biallelic pathogenic variants in the DDC gene and mainly characterized by developmental delay, hypotonia, and oculogyric crises. Early diagnosis is crucial for correct patient management; however, many patients remain misdiagnosed or undiagnosed due to the rarity and clinical heterogeneity of the disorder especially in the milder forms.
In this study exome sequencing approach by screening 2000 paediatric patients with neurodevelopmental disorders to identify possible new AADC variants and AADC deficiency patients is applied. Five distinct DDC variants in two unrelated individuals are identified. The variants are classified as benign class I variants and therefore non-causative according to the ACMG/AMP guidelines. The patients in this study carrying DDC variants present clinical manifestations not precisely overlapped to the classical symptoms exhibited by the most severe AADC deficiency cases. However, screening data derived from exome sequencing in patients featuring wide-range symptoms related to neurodevelopmental disorders may help to identify AADC deficiency patients, especially when applied to larger cohorts. Read the full article here.
 On Orphanet’s Rare Diseases and Orphan Drugs Portal (https://www.orpha.net/consor/cgi-bin/index.php) you can now use a new application in English to search for a rare disease by clinical signs or symptoms .
On Orphanet’s Rare Diseases and Orphan Drugs Portal (https://www.orpha.net/consor/cgi-bin/index.php) you can now use a new application in English to search for a rare disease by clinical signs or symptoms .
Orphanet provides a clinical description of rare diseases using a range of clinical signs and symptoms. This description, based on cases published in the biomedical literature, uses the phenotypic abnormalities specified in the Human Phenotype Ontology, HPO.
The application is available at the following link: https://clinicalsigns.orphanet.app and in the section “Clinical signs and symptoms” you can search by entering signs and symptoms that interest you. In the search bar, enter the clinical sign/symptom you want to search for and select the corresponding HPO term.
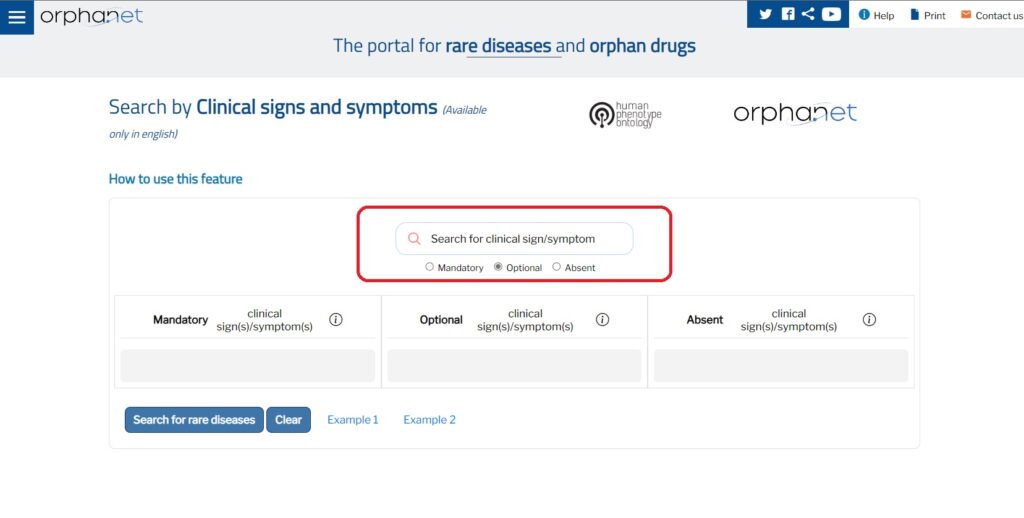
The information provided is calculated for the entire population of patients in routine medical practice.
 In the United States, rare disease is defined as a condition that affects fewer than 200,000 individuals. Collectively, rare diseases affect an estimated 30 million Americans. A significant portion of rare diseases have an underlying genetic cause; however, this may go undiagnosed. To better serve these patients, the Mayo Clinic Program for Rare and Undiagnosed Diseases was created under the auspices of the Center for Individualized Medicine aiming to integrate genomics into subspecialty practice including targeted genetic testing, research, and education.
In the United States, rare disease is defined as a condition that affects fewer than 200,000 individuals. Collectively, rare diseases affect an estimated 30 million Americans. A significant portion of rare diseases have an underlying genetic cause; however, this may go undiagnosed. To better serve these patients, the Mayo Clinic Program for Rare and Undiagnosed Diseases was created under the auspices of the Center for Individualized Medicine aiming to integrate genomics into subspecialty practice including targeted genetic testing, research, and education.
Implementation of the Program for Rare and Undiagnosed Diseases began in 2018 and Genetic Testing and Counseling unit launched in 2020 to support program expansion. Currently, 29 clinical indications for rare diseases are included in 11 specialty departments.
Implementation of Program for Rare and Undiagnosed Diseases began have enabled subspecialty practices advance expertise in rare disease where genetic counselors have not historically been embedded in practice. Democratizing access to genetic testing and counseling can broaden the reach of patients with rare disease and increase the diagnostic yield of such indications leading to better medical management as well as expanding research opportunities. Read the full article here.
 Rosai-Dorfman disease is a rare benign histiocytic disorder characterized in most cases by painless cervical adenopathy. Less than 10% of extranodal cases involve bony lesions. Primary bone Rosai-Dorfman disease in the absence of nodal disease is extremely rare.
Rosai-Dorfman disease is a rare benign histiocytic disorder characterized in most cases by painless cervical adenopathy. Less than 10% of extranodal cases involve bony lesions. Primary bone Rosai-Dorfman disease in the absence of nodal disease is extremely rare.
In this report a case of 48 year-old male presented with progressive right-sided otalgia, tinnitus, vertigo, and hearing loss is presented. A right temporal bone lytic lesion is detected on diagnostic imaging. Resection of the lesion and histopathological examination revealed Rosai-Dorfman disease.
Rosai-Dorfman disease primary bone lesions are an atypical presentation of a rare disease. This is the second reported case of Rosai-Dorfman disease arising within the temporal bone. This case study reveals that Rosai-Dorfman disease should be considered for patients presenting with inflammatory/lytic lesions of the temporal bone, in cases where infection and malignancy have been excluded. Read the full article here.
 White Sutton Syndrome is a rare autosomal dominant disorder resulting from a de novo mutation. The phenotype is characterized by a wide spectrum of cognitive dysfunction and developmental delays. Hearing loss is frequently mentioned as one of the symptoms of this rare disease, but details are usually scant. In this report a case of a male child affected by White Sutton Syndrome and sensorineural hearing loss is shown. Up to date, the present case is the first description of hearing loss due to an auditory neuropathy spectrum disorder in White Sutton Syndrome. A comprehensive audiological assessment is therefore mandatory in all White Sutton Syndrome patients in order to recognize a possible auditory neuropathy disorder and then avoid misdiagnosis, or erroneous clinical management. Read the full article here.
White Sutton Syndrome is a rare autosomal dominant disorder resulting from a de novo mutation. The phenotype is characterized by a wide spectrum of cognitive dysfunction and developmental delays. Hearing loss is frequently mentioned as one of the symptoms of this rare disease, but details are usually scant. In this report a case of a male child affected by White Sutton Syndrome and sensorineural hearing loss is shown. Up to date, the present case is the first description of hearing loss due to an auditory neuropathy spectrum disorder in White Sutton Syndrome. A comprehensive audiological assessment is therefore mandatory in all White Sutton Syndrome patients in order to recognize a possible auditory neuropathy disorder and then avoid misdiagnosis, or erroneous clinical management. Read the full article here.
 Hemiplegic migraine is a rare disease and data concerning the pediatric population are even more rare as they are often extrapolated from mixed cohorts.
Hemiplegic migraine is a rare disease and data concerning the pediatric population are even more rare as they are often extrapolated from mixed cohorts.
The aim of this study is to describe a cohort of pediatric patients with genetically confirmed familial hemiplegic migraine (FHM). The knowledge of genotype-phenotype correlations may suggest prognostic factors associated with severe phenotypes.
Patients who met International Classification of Headache Disorders, third edition criteria for FHM, who had a molecular diagnosis, and whose first attack occurred under the age of 18 years are selected.
The study data show that most of the patients with early-onset FHM experience infrequent and non-severe attacks, which improve over time. Furthermore, the clinical course reveals neither the appearance of novel neurological disorders or a deterioration of basic neurological or cognitive functioning. Read the full article here.