
 На 1-ви април стартира проектът ORPHANET DATA FOR RARE DISEASES 2. След пилотна фаза през 2022 г., той има за цел да допринесе за постигане на амбициите, поставени от Rare 2030 по отношение на данните. Въпреки факта, че номенклатурата на Orphanet вече съществува и е свободно достъпна в обработваем формат, реалното внедряването в системите за здравна информация е предизвикателство поради хетерогенността им по отношение на кодиране и практиките, както и инструментите. Опитът от проекта RD-CODE, поддържащ различни модели за внедряване в четири държави-членки, ни научи, че е необходима локална поддръжка на местен език за програмисти и технически екипи, за да се постигне правилно внедряване в съответствие с насоките за добри практики за кодиране и следователно да се повиши качеството на данните и съпоставимост. За да се отговори на тези нужди, е важно да се поддържа номенклатурата на Orphanet за редки болести и да се подобрява, като се основава на добре организирания и структуриран експертен опит в областта на редките болести, заложен в ERN, за да се увеличи нейната оперативна съвместимост с други терминологии, използвани в различни страни и в регистрите , и да допринесе за приемането и внедряването, като се започне от болниците, участващи в ERN, като се създаде мрежа от национални номенклатурни центрове на Orphanet в 19 държави-членки и накрая се улесни вземането на основано на доказателства решение чрез използване на базата от знания на Orphanet, свързана с ORPHA -кодовете. Прочетете повече тук.
На 1-ви април стартира проектът ORPHANET DATA FOR RARE DISEASES 2. След пилотна фаза през 2022 г., той има за цел да допринесе за постигане на амбициите, поставени от Rare 2030 по отношение на данните. Въпреки факта, че номенклатурата на Orphanet вече съществува и е свободно достъпна в обработваем формат, реалното внедряването в системите за здравна информация е предизвикателство поради хетерогенността им по отношение на кодиране и практиките, както и инструментите. Опитът от проекта RD-CODE, поддържащ различни модели за внедряване в четири държави-членки, ни научи, че е необходима локална поддръжка на местен език за програмисти и технически екипи, за да се постигне правилно внедряване в съответствие с насоките за добри практики за кодиране и следователно да се повиши качеството на данните и съпоставимост. За да се отговори на тези нужди, е важно да се поддържа номенклатурата на Orphanet за редки болести и да се подобрява, като се основава на добре организирания и структуриран експертен опит в областта на редките болести, заложен в ERN, за да се увеличи нейната оперативна съвместимост с други терминологии, използвани в различни страни и в регистрите , и да допринесе за приемането и внедряването, като се започне от болниците, участващи в ERN, като се създаде мрежа от национални номенклатурни центрове на Orphanet в 19 държави-членки и накрая се улесни вземането на основано на доказателства решение чрез използване на базата от знания на Orphanet, свързана с ORPHA -кодовете. Прочетете повече тук.
 Синдромът на Phelan-McDermid (PMS) е рядко генетично състояние, причинено от делеция, обхващаща региона 22q13.3 или патогенен вариант на гена SHANK3. Клиничното представяне е променливо, но основните характеристики включват глобално изоставане в развитието/интелектуална недостатъчност, изразено увреждане или забавяне на речта, заедно с други характеристики като хипотония и соматични или психиатрични съпътстващи заболявания. Този доклад очертава темите за психичното здраве, развитието и поведението през целия живот на хората с PMS, както е съобщено от родители/болногледачи, експерти и други ключови специалисти, участващи в грижите за PMS. В този доклад са представени няколко препоръки, базирани на наличната литература относно психичното здраве и поведение при PMS. Освен това тази статия има за цел да подобри осъзнаването на значението на отчитането на нивото на развитие на хора с PMS при оценката на психичното здраве и поведенчески проблеми. Разбирането как несъответствието между нивото на развитие и календарната възраст може да повлияе на поведението предлага представа за значението на това и дава информация за грижите за лица с PMS, което позволява на клиницистите да се справят с незадоволените грижи (за психично здраве) и да подобрят качеството на живот на тези пациенти. Прочетете цялата статия тук.
Синдромът на Phelan-McDermid (PMS) е рядко генетично състояние, причинено от делеция, обхващаща региона 22q13.3 или патогенен вариант на гена SHANK3. Клиничното представяне е променливо, но основните характеристики включват глобално изоставане в развитието/интелектуална недостатъчност, изразено увреждане или забавяне на речта, заедно с други характеристики като хипотония и соматични или психиатрични съпътстващи заболявания. Този доклад очертава темите за психичното здраве, развитието и поведението през целия живот на хората с PMS, както е съобщено от родители/болногледачи, експерти и други ключови специалисти, участващи в грижите за PMS. В този доклад са представени няколко препоръки, базирани на наличната литература относно психичното здраве и поведение при PMS. Освен това тази статия има за цел да подобри осъзнаването на значението на отчитането на нивото на развитие на хора с PMS при оценката на психичното здраве и поведенчески проблеми. Разбирането как несъответствието между нивото на развитие и календарната възраст може да повлияе на поведението предлага представа за значението на това и дава информация за грижите за лица с PMS, което позволява на клиницистите да се справят с незадоволените грижи (за психично здраве) и да подобрят качеството на живот на тези пациенти. Прочетете цялата статия тук.
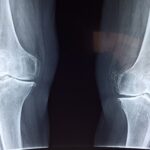 Множествената епифизарна дисплазия, която засяга епифизите на дългите кости, може да има автозомно-доминантни и автозомно-рецесивни модели на унаследяване. Признаците обикновено се появяват в детството, въпреки че понякога симптомите не са налични до зряла възраст. Целите на лечението при деца са да се предотврати ранната поява на остеоартрит, да се подобри функцията и да се обучат пациентите и техните семейства относно естествената история и генетичната основа на заболяването. Някои пациенти идват в клиниката само с нелекуваща се и неидентифицирана болка в ставите. Въпреки че множествената епифизарна дисплазия тип 5 е рядко заболяване с автозомно доминантно унаследяване, тя може да се наблюдава и при de novo мутация, макар и много рядко, без фамилна анамнеза.
Множествената епифизарна дисплазия, която засяга епифизите на дългите кости, може да има автозомно-доминантни и автозомно-рецесивни модели на унаследяване. Признаците обикновено се появяват в детството, въпреки че понякога симптомите не са налични до зряла възраст. Целите на лечението при деца са да се предотврати ранната поява на остеоартрит, да се подобри функцията и да се обучат пациентите и техните семейства относно естествената история и генетичната основа на заболяването. Някои пациенти идват в клиниката само с нелекуваща се и неидентифицирана болка в ставите. Въпреки че множествената епифизарна дисплазия тип 5 е рядко заболяване с автозомно доминантно унаследяване, тя може да се наблюдава и при de novo мутация, макар и много рядко, без фамилна анамнеза.
Пациент от мъжки пол на 7 години постъпи в амбулатория по ортопедия с оплаквания от болки в ставите от около 3 години, отпадналост, болки в коленете и глезените. В лабораторните резултати няма очевидна находка, освен дефицит на витамин D. Епифизите, особено в глезена, са диспластични на рентгенова снимка. След генетични изследвания е открита множествена епифизарна дисплазия тип 5, с de novo мутация, без фамилна история.
Множествената епифизарна дисплазия тип 5 е рядко заболяване. Трябва да се има предвид, че скелетната дисплазия също трябва да бъде оценена, въпреки че рядко се наблюдава при пациенти с постоянна болка в ставите. По този начин е възможно да се забави прогресията с ранна диагностика на пациента и да се сведат до минимум ранните хирургични изисквания. Прочетете цялата статия тук.
 Нововъзникващите технологии за машинно самообучение (МС) имат потенциала значително да подобрят изследванията и лечението на редки заболявания, които представляват огромен набор от заболявания, засягащи малка част от цялото население. Алгоритмите за изкуствен интелект (ИИ) могат да помогнат за бързото идентифициране на модели и асоциации, които биха били трудни или невъзможни за откриване от човешки анализатори. Техниките за прогнозно моделиране като дълбоко обучение са използвани за прогнозиране на прогресията на редки заболявания, което позволява разработването на по-целенасочени лечения. Нещо повече, ИИ също показа обещаващи резултати в областта на разработването на лекарства за редки заболявания с идентифицирането на субпопулации от пациенти, които е най-вероятно да отговорят на определено лекарство. Този доклад има за цел да подчертае постиженията на ИИ – алгоритмите в изследването на редки заболявания през последното десетилетие и да даде съвет на изследователите кои методи са се доказали като най-ефективни. Прегледът ще се съсредоточи върху специфични редки заболявания, както е определена заболеваемост, която не надвишава 1-9/100 000 в Orphanet, и ще разгледа кои методи на ИИ са били най-успешни в тяхното изследване. Прочетете цялата статия тук.
Нововъзникващите технологии за машинно самообучение (МС) имат потенциала значително да подобрят изследванията и лечението на редки заболявания, които представляват огромен набор от заболявания, засягащи малка част от цялото население. Алгоритмите за изкуствен интелект (ИИ) могат да помогнат за бързото идентифициране на модели и асоциации, които биха били трудни или невъзможни за откриване от човешки анализатори. Техниките за прогнозно моделиране като дълбоко обучение са използвани за прогнозиране на прогресията на редки заболявания, което позволява разработването на по-целенасочени лечения. Нещо повече, ИИ също показа обещаващи резултати в областта на разработването на лекарства за редки заболявания с идентифицирането на субпопулации от пациенти, които е най-вероятно да отговорят на определено лекарство. Този доклад има за цел да подчертае постиженията на ИИ – алгоритмите в изследването на редки заболявания през последното десетилетие и да даде съвет на изследователите кои методи са се доказали като най-ефективни. Прегледът ще се съсредоточи върху специфични редки заболявания, както е определена заболеваемост, която не надвишава 1-9/100 000 в Orphanet, и ще разгледа кои методи на ИИ са били най-успешни в тяхното изследване. Прочетете цялата статия тук.
 Нововъзникнал рефрактерен епилептичен статус (NORSE), включително неговия подтип с предшестващо фебрилно състояние, известно като синдром на епилепсия, свързана с фебрилна инфекция (FIRES), е една от най-тежките форми на епилептичен статус. Точните причини за NORSE понастоящем не са известни и досега няма специфична терапия за заболяването. Идентифицирането на основната патофизиология и откриването на специфични биомаркери, независимо дали са имунологични, инфекциозни, генетични или други, може да помогне на лекарите при лечението на пациенти с NORSE. Предложен е широк спектър от биомаркери за пациенти с епилептичен статус, някои от които са оценени за пациенти с NORSE. Независимо от това, нито един не е валидиран поради значителни вариации в кохортите на изследването, събраните биопроби, приложените аналитични методи и дефинираните крайни точки на резултатите, както и малките размери на пробите. Институтът NORSE създаде отворено биохранилище NORSE/FIRES за свързани със здравето данни и биологични проби, което позволява събирането на биообразци по целия свят, насърчавайки многоцентрово изследване и споделяне на данни и проби. В този доклад са предложени стандартни оперативни процедури за събиране на биопроби и биобанкиране при това рядко състояние. Предложени са и критерии за подходящо използване на предварително събрани биопроби. Предвижда се, че широкото използване на стандартизирани процедури ще намали хетерогенността, ще улесни бъдещата идентификация на валидирани биомаркери за NORSE и ще осигури по-добро разбиране на патофизиологията и най-добрите клинични грижи за тези пациенти. Прочетете цялата статия тук.
Нововъзникнал рефрактерен епилептичен статус (NORSE), включително неговия подтип с предшестващо фебрилно състояние, известно като синдром на епилепсия, свързана с фебрилна инфекция (FIRES), е една от най-тежките форми на епилептичен статус. Точните причини за NORSE понастоящем не са известни и досега няма специфична терапия за заболяването. Идентифицирането на основната патофизиология и откриването на специфични биомаркери, независимо дали са имунологични, инфекциозни, генетични или други, може да помогне на лекарите при лечението на пациенти с NORSE. Предложен е широк спектър от биомаркери за пациенти с епилептичен статус, някои от които са оценени за пациенти с NORSE. Независимо от това, нито един не е валидиран поради значителни вариации в кохортите на изследването, събраните биопроби, приложените аналитични методи и дефинираните крайни точки на резултатите, както и малките размери на пробите. Институтът NORSE създаде отворено биохранилище NORSE/FIRES за свързани със здравето данни и биологични проби, което позволява събирането на биообразци по целия свят, насърчавайки многоцентрово изследване и споделяне на данни и проби. В този доклад са предложени стандартни оперативни процедури за събиране на биопроби и биобанкиране при това рядко състояние. Предложени са и критерии за подходящо използване на предварително събрани биопроби. Предвижда се, че широкото използване на стандартизирани процедури ще намали хетерогенността, ще улесни бъдещата идентификация на валидирани биомаркери за NORSE и ще осигури по-добро разбиране на патофизиологията и най-добрите клинични грижи за тези пациенти. Прочетете цялата статия тук.
 Традиционните клинични изпитвания изискват тестове и процедури, които се прилагат в централизирани центрове за клинични изпитвания, които са извън стандарта на грижи, които пациентите получават за техните редки и хронични заболявания. Ограниченият брой пациенти с редки заболявания, разпръснати по целия свят, прави особено предизвикателство набирането на участници и провеждането на тези традиционни клинични изпитвания.
Традиционните клинични изпитвания изискват тестове и процедури, които се прилагат в централизирани центрове за клинични изпитвания, които са извън стандарта на грижи, които пациентите получават за техните редки и хронични заболявания. Ограниченият брой пациенти с редки заболявания, разпръснати по целия свят, прави особено предизвикателство набирането на участници и провеждането на тези традиционни клинични изпитвания.
Участието в клинични изследвания може да бъде натоварващо, особено за деца, възрастни хора, лица с физически и когнитивни увреждания, които се нуждаят от транспорт и помощ от болногледач, или пациенти, които живеят в отдалечени места или не могат да си позволят транспорт. През последните години има нарастваща необходимост да се разглеждат децентрализираните клинични изпитвания като ориентиран към участниците подход, който използва нови технологии и иновативни процедури за взаимодействие с участниците в комфорта на техния дом.
Този доклад обсъжда планирането и провеждането на децентрализирани клинични изпитвания, които могат да повишат качеството на изпитванията със специфичен фокус върху редките заболявания. Прочетете цялата статия тук.
 CLN3 болестта, причинена от биалелни мутации в гена CLN3, е рядко педиатрично невродегенеративно заболяване, което няма лечение или терапия контролираща болеста. Разработването на ефективни лечения е възпрепятствано от липсата на етиологични познания, но генното заместване се очертава като обещаващ терапевтичен метод за такива разстройства. В това изследване се използва миши модел на болестта CLN3 за тестване на безопасността и ефикасността на доставяна чрез цереброспинална течност AAV9 генна терапия с дизайн на изследване, оптимизиран за превенция. В този модел постнаталното приложение на първия ден на вируса за генна терапия води до силна експресия на човешки CLN3 в цялата централна нервна система за 24-месечната продължителност на изследването. Изследват се набор от хистопатологични и поведенчески параметри, като терапията последователно и значително намалява редица характерни симптоми на заболяването, като същевременно е безопасно и се понася добре. Резултатите показват голяма перспектива за пренасяне на терапията в клиничната практика, което подтиква стартирането на първото клинично изпитване при хора. Прочетете цялата статия тук.
CLN3 болестта, причинена от биалелни мутации в гена CLN3, е рядко педиатрично невродегенеративно заболяване, което няма лечение или терапия контролираща болеста. Разработването на ефективни лечения е възпрепятствано от липсата на етиологични познания, но генното заместване се очертава като обещаващ терапевтичен метод за такива разстройства. В това изследване се използва миши модел на болестта CLN3 за тестване на безопасността и ефикасността на доставяна чрез цереброспинална течност AAV9 генна терапия с дизайн на изследване, оптимизиран за превенция. В този модел постнаталното приложение на първия ден на вируса за генна терапия води до силна експресия на човешки CLN3 в цялата централна нервна система за 24-месечната продължителност на изследването. Изследват се набор от хистопатологични и поведенчески параметри, като терапията последователно и значително намалява редица характерни симптоми на заболяването, като същевременно е безопасно и се понася добре. Резултатите показват голяма перспектива за пренасяне на терапията в клиничната практика, което подтиква стартирането на първото клинично изпитване при хора. Прочетете цялата статия тук.
 Придобитата Хемофилия А е рядко заболяване с годишна заболеваемост от 1,48 на милион. Въз основа на клинични наблюдения, в тази статия се подозира по-висока заболеваемост в южна Швейцария и нейната цел е да предостави местни епидемиологични данни и клинична информация относно диагнозата, лечението и изхода в този регион. Всички възрастни пациенти с придобита Хемофилия А, лекувани между 2013 и 2019 г. в лечебно заведение в южна Швейцария, са включени в настоящия ретроспективен анализ.
Придобитата Хемофилия А е рядко заболяване с годишна заболеваемост от 1,48 на милион. Въз основа на клинични наблюдения, в тази статия се подозира по-висока заболеваемост в южна Швейцария и нейната цел е да предостави местни епидемиологични данни и клинична информация относно диагнозата, лечението и изхода в този регион. Всички възрастни пациенти с придобита Хемофилия А, лекувани между 2013 и 2019 г. в лечебно заведение в южна Швейцария, са включени в настоящия ретроспективен анализ.
11 пациенти с придобита Хемофилия А между 2013 г. и 2019 г. са били лекувани, което ознчава годишна заболеваемост от 4,5 на милион. Средната продължителност от първите симптоми до диагнозата е 4,5 дни, а средната възраст при поставяне на диагнозата е 79 години. Възможните причинни състояния са: бременност, нодозен полиартериит, миелодиспластичен синдром, хроничен човешки имунодефицитен вирус (ХИВ) и постекспозиционна профилактика на ХИВ. При пет пациенти не е идентифицирано основно или свързано състояние. Всички пациенти са имали симптоми на кървене, 5/10 пациенти са имали големи кръвоизливи и 7/10 пациенти са били лекувани с байпасни средства. Всички пациенти са получавали кортикостероиди; 7/10 пациенти са получили имуносупресивна комбинирана терапия.
Придобитата Хемофилия А е рядко заболяване, но лечимо въпреки напредналата възраст на пациента и съпътстващите заболявания. Честотата му в южна Швейцария е по-висока, отколкото се предполагаше преди. Прочетете цялата статия тук.
![]() За 14-та поредна година предстои да се проведе Национална конференция за редки болести и лекарства сираци, организирана от Институт по редки болести. Събитието е ежегоден форум, който събира заедно всички заинтересовани страни – медицински специалисти, пациенти, студенти по медицина, представители на индустрията и здравни власти, за да представят и обсъдят новостите в диагностиката, лечението и проследяването на редките болести, развитието на европейските референтни мрежи и достъпа до иновации.
За 14-та поредна година предстои да се проведе Национална конференция за редки болести и лекарства сираци, организирана от Институт по редки болести. Събитието е ежегоден форум, който събира заедно всички заинтересовани страни – медицински специалисти, пациенти, студенти по медицина, представители на индустрията и здравни власти, за да представят и обсъдят новостите в диагностиката, лечението и проследяването на редките болести, развитието на европейските референтни мрежи и достъпа до иновации.
Място: хотел „Империал” – гр. Пловдив
Краен срок за ранна регистрация: 1 август 2023
Краен срок за късна регистрация: 15 септември 2023
Краен срок за изпращане на резюмета: 1 септември 2023
За контакти с организационния комитет: congress@raredis.org
Допълнителнa информация можете да намерите на XIV НАЦИОНАЛНА КОНФЕРЕНЦИЯ ЗА РЕДКИ БОЛЕСТИ И ЛЕКАРСТВА СИРАЦИ – Виртуален Конгресен Център (raredis.org)
 Този доклад предоставя изчерпателно обобщение на доказателства за текущата ситуация с редките болести (РБ) в световен и регионален мащаб, включително условия, практики, политики и разпоредби, както и предизвикателствата и бариерите, пред които са изправени пациентите с РБ, техните семейства и лицата, които се грижат за тях. Документът се основава на преглед на академична литература, политики и процес на валидиране и обратна връзка от група от седем експерти от цял свят. Докладът е разделен на пет основни раздела: методология и цел; фон и контекст; преглед на текущата ситуация и ключовите предизвикателства, свързани с РБ, обхващащи шест измерения: тежест на заболяването, пътя на пациента, социално въздействие, управление на заболяването, политики, свързани с РБ, и научноизследователска и развойна дейност; препоръки; заключения. Препоръките произлизат от дискусията, предприета от експертите относно констатациите от този преглед, и предоставят набор от приложими решения на предизвикателствата и пречките пред подобряването на достъпа до диагностика и лечение на РБ по света. Препоръките могат да подкрепят вземането на критични решения, насочвайки усилията на широк кръг от заинтересовани страни от РБ, включително правителства, международни организации, производители, изследователи и организации за защита на пациентите. Прочетете цялата статия тук.
Този доклад предоставя изчерпателно обобщение на доказателства за текущата ситуация с редките болести (РБ) в световен и регионален мащаб, включително условия, практики, политики и разпоредби, както и предизвикателствата и бариерите, пред които са изправени пациентите с РБ, техните семейства и лицата, които се грижат за тях. Документът се основава на преглед на академична литература, политики и процес на валидиране и обратна връзка от група от седем експерти от цял свят. Докладът е разделен на пет основни раздела: методология и цел; фон и контекст; преглед на текущата ситуация и ключовите предизвикателства, свързани с РБ, обхващащи шест измерения: тежест на заболяването, пътя на пациента, социално въздействие, управление на заболяването, политики, свързани с РБ, и научноизследователска и развойна дейност; препоръки; заключения. Препоръките произлизат от дискусията, предприета от експертите относно констатациите от този преглед, и предоставят набор от приложими решения на предизвикателствата и пречките пред подобряването на достъпа до диагностика и лечение на РБ по света. Препоръките могат да подкрепят вземането на критични решения, насочвайки усилията на широк кръг от заинтересовани страни от РБ, включително правителства, международни организации, производители, изследователи и организации за защита на пациентите. Прочетете цялата статия тук.