
 The manifestations of Phelan-McDermid syndrome (PMS) are complex, warranting expert and multidisciplinary care in all life stages. In this report consensus recommendations on the organization of care for individuals with PMS are proposed. It is indicated that care should consider all life domains, which can be done within the framework of the International Classification of Functioning, Disability and Health (ICF). This framework assesses disability and functioning as the outcome of the individual’s interactions with other factors. The different roles within care, such as performed by a centre of expertise, by regional health care providers and by a coordinating physician are addressed. A surveillance scheme and emergency card is provided and disciplines participating in a multidisciplinary team for PMS are described. Additionally, recommendations are provided for transition from paediatric to adult care. This care proposition may also be useful for individuals with other rare genetic neurodevelopmental disorders. Read the full article here.
The manifestations of Phelan-McDermid syndrome (PMS) are complex, warranting expert and multidisciplinary care in all life stages. In this report consensus recommendations on the organization of care for individuals with PMS are proposed. It is indicated that care should consider all life domains, which can be done within the framework of the International Classification of Functioning, Disability and Health (ICF). This framework assesses disability and functioning as the outcome of the individual’s interactions with other factors. The different roles within care, such as performed by a centre of expertise, by regional health care providers and by a coordinating physician are addressed. A surveillance scheme and emergency card is provided and disciplines participating in a multidisciplinary team for PMS are described. Additionally, recommendations are provided for transition from paediatric to adult care. This care proposition may also be useful for individuals with other rare genetic neurodevelopmental disorders. Read the full article here.
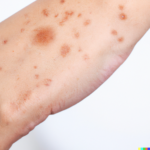 Sweet syndrome (SS) is a rare disease described as a febrile neutrophilic dermatosis with acute onset, the pathogenesis of which has not yet been elucidated. The syndrome is characterized by the sudden onset of erythematous infiltrated papules or plaques located on the upper body and is associated with fever, leukocytosis and neutrophilia. The lesions show a dense dermal infiltration with mature neutrophils. The condition is responsive to systemic steroids. The central nervous system, bones, muscles, eyes, ears, mouth, heart, lung, liver, kidneys, intestines, and spleen may be affected by SS as extracutaneous manifestations. Up to 80% of SS cases are associated with hematological diseases, predominantly myelodysplastic syndrome (MDS). Myelodysplastic syndrome is a clonal disease of the bone marrow characterized by inefficient hematopoiesis, dysplasia of the bone marrow and peripheral cytopenias. After analyzing later studies and current practical aspects regarding MDS-related SS, in this report an algorithm for evaluating these patients is suggested. Read the full article here.
Sweet syndrome (SS) is a rare disease described as a febrile neutrophilic dermatosis with acute onset, the pathogenesis of which has not yet been elucidated. The syndrome is characterized by the sudden onset of erythematous infiltrated papules or plaques located on the upper body and is associated with fever, leukocytosis and neutrophilia. The lesions show a dense dermal infiltration with mature neutrophils. The condition is responsive to systemic steroids. The central nervous system, bones, muscles, eyes, ears, mouth, heart, lung, liver, kidneys, intestines, and spleen may be affected by SS as extracutaneous manifestations. Up to 80% of SS cases are associated with hematological diseases, predominantly myelodysplastic syndrome (MDS). Myelodysplastic syndrome is a clonal disease of the bone marrow characterized by inefficient hematopoiesis, dysplasia of the bone marrow and peripheral cytopenias. After analyzing later studies and current practical aspects regarding MDS-related SS, in this report an algorithm for evaluating these patients is suggested. Read the full article here.
 Idiopathic systemic capillary leak syndrome (ISCLS) is a rare disease characterized by recurrent episodes of acute life-threatening attacks of shock, hemoconcentration, and hypoalbuminemia. Increase in capillary permeability results in reversible plasma movement into the interstitial spaces followed by appearance of related symptoms or complications, including renal failure. This condition can be potentially life-threatening; however, it is easily misdiagnosed.
Idiopathic systemic capillary leak syndrome (ISCLS) is a rare disease characterized by recurrent episodes of acute life-threatening attacks of shock, hemoconcentration, and hypoalbuminemia. Increase in capillary permeability results in reversible plasma movement into the interstitial spaces followed by appearance of related symptoms or complications, including renal failure. This condition can be potentially life-threatening; however, it is easily misdiagnosed.
A 47-year-old man with no previous medical history presented to the emergency department after experiencing general weakness and abdominal pain. He develops hypovolemic shock within 3 h of presentation and initial laboratory tests showed hemoconcentration, hypoalbuminemia and acute kidney injury. Following vigorous fluid therapy and supportive care, the patient recovers, but a similar episode recurrs after 4 months without any specific trigger. Based on the combined clinical manifestations and laboratory findings of both the attacks, he is diagnosed with ISCLS. Symptomatic relief is achieved via oxygen supplementation and massive volume replacement using normal saline and the patient is prescribed bambuterol 10 mg and theophylline 400 mg once-a-day. He is discharged from the hospital on day 5 of hospitalization. Thereafter, the patient is being followed for 5 years without any symptoms or recurrence of ISCLS even in the situation of COVID-19 infection.
ISCLS is an extremely infrequent and commonly misdiagnosed disease. However, early diagnosis, treatment and prophylaxis through accumulated clinical data can prevent ISCLS recurrence and the development of related fatal complications. Therefore, clinicians need to be well aware of the variety of clinical characteristics and treatment options of this disease. Read the full article here.
 Advances in the study of ultra-rare genetic conditions are leading to the development of targeted interventions developed for single or very small numbers of patients. Due to the experimental but also highly individualized nature of these interventions, they are difficult to classify cleanly as either research or clinical care. The goal of this report is to understand how parents, IRB members, and clinical geneticists familiar with individualized genetic interventions conceptualize these activities and their implications for the relationship between research and clinical care.
Advances in the study of ultra-rare genetic conditions are leading to the development of targeted interventions developed for single or very small numbers of patients. Due to the experimental but also highly individualized nature of these interventions, they are difficult to classify cleanly as either research or clinical care. The goal of this report is to understand how parents, IRB members, and clinical geneticists familiar with individualized genetic interventions conceptualize these activities and their implications for the relationship between research and clinical care.
In this report, qualitative, semi-structured interviews with 28 parents, IRB members, and clinical geneticists are conducted, and themes from those interviews through content analysis are derived.
Individualized interventions aimed at one or few patients reveal the limitations of a binary framing of research and clinical care. As a hybrid set of activities, individualized interventions suggest the need for flexibility and new frameworks that acknowledge these activities across the spectrum of research and clinical care. Read the full article here.
 Kikuchi disease (KD) is a benign self-limiting rare disease with unknown etiology. Prolonged fever with tender neck lymphadenitis is the most common presentation. Blood tests are not specific, and the final diagnosis is by biopsy. In this report two patients, ages seven and twelve years, who present with fever and neck lymphadenitis, are described. Both cases receive antibiotics for more than two weeks without improvement. Blood work shows high inflammatory markers. The manifestation of the second case overlaps with Hashimoto’s disease. The later diagnosis was confirmed by lymph node (LN) biopsy. Read the full article here.
Kikuchi disease (KD) is a benign self-limiting rare disease with unknown etiology. Prolonged fever with tender neck lymphadenitis is the most common presentation. Blood tests are not specific, and the final diagnosis is by biopsy. In this report two patients, ages seven and twelve years, who present with fever and neck lymphadenitis, are described. Both cases receive antibiotics for more than two weeks without improvement. Blood work shows high inflammatory markers. The manifestation of the second case overlaps with Hashimoto’s disease. The later diagnosis was confirmed by lymph node (LN) biopsy. Read the full article here.
 Henoch-Schönlein purpura (HSP) is the most common vasculitis in childhood, presenting with purpura, predominantly of the lower extremities and occasionally with renal involvement as well. Although associated with childhood, HSP, although rarely, can also develop in adults as well. It this report a patient in his sixties is presented, presenting with a myriad of rash units on his lower extremities, including bullous ones, and a constellation of chronic kidney failure. Skin and renal biopsy specimens reveal morphological changes and immune depositions representative of HSP. Despite treatment, the patient’s kidney failure slowly progresses. Although rare, the bullous form of HSP can be viewed as a more aggressive form of the disease, as seen by the presentation constellation and rapid progression in this case. Read the full article here.
Henoch-Schönlein purpura (HSP) is the most common vasculitis in childhood, presenting with purpura, predominantly of the lower extremities and occasionally with renal involvement as well. Although associated with childhood, HSP, although rarely, can also develop in adults as well. It this report a patient in his sixties is presented, presenting with a myriad of rash units on his lower extremities, including bullous ones, and a constellation of chronic kidney failure. Skin and renal biopsy specimens reveal morphological changes and immune depositions representative of HSP. Despite treatment, the patient’s kidney failure slowly progresses. Although rare, the bullous form of HSP can be viewed as a more aggressive form of the disease, as seen by the presentation constellation and rapid progression in this case. Read the full article here.
 Myelin oligodendrocyte glycoprotein (MOG)-associated disease (MOGAD) is a rare, antibody-mediated inflammatory demyelinating disorder of the central nervous system (CNS) that has varying phenotypes. A rare case of a nine-year-old girl who presents with a drop in her academic performance and right-sided Epilepsia partialis continua is reported. Magnetic resonance imaging (MRI) of the brain detected evidence for unilateral (left) cortical encephalitis with juxtacortical edema. An electroencephalogram revealed a hemi-generalized poly spike and wave discharges in the left hemisphere, several of which correlated with myoclonic jerks. The cerebrospinal fluid (CSF) analysis was normal. Autoimmune workup resulted in a positive serum MOG-immunoglobulin G (IgG), which confirmed the diagnosis. The child showed an excellent clinical response to intravenous methylprednisolone and intravenous immunoglobulins therapy. Read the full article here.
Myelin oligodendrocyte glycoprotein (MOG)-associated disease (MOGAD) is a rare, antibody-mediated inflammatory demyelinating disorder of the central nervous system (CNS) that has varying phenotypes. A rare case of a nine-year-old girl who presents with a drop in her academic performance and right-sided Epilepsia partialis continua is reported. Magnetic resonance imaging (MRI) of the brain detected evidence for unilateral (left) cortical encephalitis with juxtacortical edema. An electroencephalogram revealed a hemi-generalized poly spike and wave discharges in the left hemisphere, several of which correlated with myoclonic jerks. The cerebrospinal fluid (CSF) analysis was normal. Autoimmune workup resulted in a positive serum MOG-immunoglobulin G (IgG), which confirmed the diagnosis. The child showed an excellent clinical response to intravenous methylprednisolone and intravenous immunoglobulins therapy. Read the full article here.
 Kenny-Caffey syndrome (KCS) is a rare hereditary disorder characterized by short stature, hypoparathyroidism and electrolyte disturbances. Kenny-Caffey syndrome type 1 and Kenny-Caffey syndrome type 2 are caused by pathogenic variants in TBCE and FAM111A, respectively. Clinically the phenotypes are difficult to distinguish.
Kenny-Caffey syndrome (KCS) is a rare hereditary disorder characterized by short stature, hypoparathyroidism and electrolyte disturbances. Kenny-Caffey syndrome type 1 and Kenny-Caffey syndrome type 2 are caused by pathogenic variants in TBCE and FAM111A, respectively. Clinically the phenotypes are difficult to distinguish.
The objective of this study is to determine and expand the phenotypic spectrum of Kenny-Caffey syndrome type 1 and Kenny-Caffey syndrome type 2 in order to anticipate on complications that may arise in these disorders.
In this study clinically and genetically ten Kenny-Caffey syndrome type 2 patients from seven families are analyzed. Because unusual phenotypes in this study‘s cohort are found, a systematic review of genetically confirmed Kenny-Caffey syndrome cases using PubMed and Scopus is performed.
This study’s case series establishes chronic kidney disease as a new feature of Kenny-Caffey syndrome type 2. In literature, substantial overlap in the phenotypic spectra of Kenny-Caffey syndrome type 1 and Kenny-Caffey syndrome type 2 is established, but identifying intellectual disability and the abnormal bone phenotype are the most distinguishing features. Read the full article here.
 Stevens-Johnson Syndrome (SJS) and toxic epidermal necrolysis (TEN) are serious and rare diseases, most often drug-induced, and their incidence has been estimated at 6 cases/million/year in France. SJS and TEN belong to the same spectrum of disease known as epidermal necrolysis (EN). They are characterized by more or less extensive epidermal detachment, associated with mucous membrane involvement, and may be complicated during the acute phase by fatal multiorgan failure. SJS and TEN can lead to severe ophthalmologic sequelae. There are no recommendations for ocular management during the chronic phase. A national audit of current practice in the 11 sites of the French reference center for toxic bullous dermatoses and a review of the literature is conducted to establish therapeutic consensus guidelines. Ophthalmologists and dermatologists from the French reference center for epidermal necrolysis are asked to complete a questionnaire on management practices in the chronic phase of SJS/TEN. Based on the results from the survey and literature review, an evaluation form to facilitate ophthalmic data collection in the chronic phase of EN is proposed and also an algorithm for the ophthalmologic management of ocular sequelae is recommended. Read the full article here.
Stevens-Johnson Syndrome (SJS) and toxic epidermal necrolysis (TEN) are serious and rare diseases, most often drug-induced, and their incidence has been estimated at 6 cases/million/year in France. SJS and TEN belong to the same spectrum of disease known as epidermal necrolysis (EN). They are characterized by more or less extensive epidermal detachment, associated with mucous membrane involvement, and may be complicated during the acute phase by fatal multiorgan failure. SJS and TEN can lead to severe ophthalmologic sequelae. There are no recommendations for ocular management during the chronic phase. A national audit of current practice in the 11 sites of the French reference center for toxic bullous dermatoses and a review of the literature is conducted to establish therapeutic consensus guidelines. Ophthalmologists and dermatologists from the French reference center for epidermal necrolysis are asked to complete a questionnaire on management practices in the chronic phase of SJS/TEN. Based on the results from the survey and literature review, an evaluation form to facilitate ophthalmic data collection in the chronic phase of EN is proposed and also an algorithm for the ophthalmologic management of ocular sequelae is recommended. Read the full article here.
 Spinocerebellar ataxia type 11 (SCA11) is a rare type of autosomal dominant cerebellar ataxia, mainly characterized by progressive cerebellar ataxia, abnormal eye signs and dysarthria. SCA11 is caused by variants in TTBK2, which encodes tau tubulin kinase 2 (TTBK2) protein. Only a few families with SCA11 were described to date, all harbouring small deletions or insertions that result in frameshifts and truncated TTBK2 proteins. In addition, TTBK2 missense variants were also reported but they were either benign or still needed functional validation to ascertain their pathogenic potential in SCA11. The mechanisms behind cerebellar neurodegeneration mediated by TTBK2 pathogenic alleles are not clearly established. There is only one neuropathological report and a few functional studies in cell or animal models published to date. Moreover, it is still unclear whether the disease is caused by TTBK2 haploinsufficiency of by a dominant negative effect of TTBK2 truncated forms on the normal allele. Although TTBK2 has a proven function in cilia formation, the phenotype caused by heterozygous TTBK2 truncating variants are not clearly typical of ciliopathies. Thus, other cellular mechanisms may explain the phenotype seen in SCA11. Read the full article here.
Spinocerebellar ataxia type 11 (SCA11) is a rare type of autosomal dominant cerebellar ataxia, mainly characterized by progressive cerebellar ataxia, abnormal eye signs and dysarthria. SCA11 is caused by variants in TTBK2, which encodes tau tubulin kinase 2 (TTBK2) protein. Only a few families with SCA11 were described to date, all harbouring small deletions or insertions that result in frameshifts and truncated TTBK2 proteins. In addition, TTBK2 missense variants were also reported but they were either benign or still needed functional validation to ascertain their pathogenic potential in SCA11. The mechanisms behind cerebellar neurodegeneration mediated by TTBK2 pathogenic alleles are not clearly established. There is only one neuropathological report and a few functional studies in cell or animal models published to date. Moreover, it is still unclear whether the disease is caused by TTBK2 haploinsufficiency of by a dominant negative effect of TTBK2 truncated forms on the normal allele. Although TTBK2 has a proven function in cilia formation, the phenotype caused by heterozygous TTBK2 truncating variants are not clearly typical of ciliopathies. Thus, other cellular mechanisms may explain the phenotype seen in SCA11. Read the full article here.