
 Impacted tooth is one that has not fully or partially erupted and is “retained” in its eruption, remaining in the bone in the wrong position against another tooth, bone, or soft tissue, so that further eruption is unlikely. This condition is most common in third molars and is thought to be found in about 73% of young adults in Europe. Dentigerous cyst is the second most common odontogenic cyst and represents about 20-24% of all odontogenic cysts of the jaws. These cysts usually remain asymptomatic and rarely reach large sizes and displace the associated tooth. In our article we present four cases of impacted third molars with atypical localization and concomitant large dentigerous cysts. A brief review of the literature with similar findings is presented, as well as the different approaches to the treatment of these cases. For more information click here.
Impacted tooth is one that has not fully or partially erupted and is “retained” in its eruption, remaining in the bone in the wrong position against another tooth, bone, or soft tissue, so that further eruption is unlikely. This condition is most common in third molars and is thought to be found in about 73% of young adults in Europe. Dentigerous cyst is the second most common odontogenic cyst and represents about 20-24% of all odontogenic cysts of the jaws. These cysts usually remain asymptomatic and rarely reach large sizes and displace the associated tooth. In our article we present four cases of impacted third molars with atypical localization and concomitant large dentigerous cysts. A brief review of the literature with similar findings is presented, as well as the different approaches to the treatment of these cases. For more information click here.
We present a case of a male patient admitted for emergency treatment at the Clinic for Maxillofacial Surgery of the University Hospital “St. George” in Plovdiv with suspected lower jaw fracture. The patient had previous nephrectomy for renal cancer in 2016. Bone metastases were detected in 2017 and therapy with Zometa was undertaken. In 2018 the patient was diagnosed with osteonecrosis of the mandible. The patient reports that he has been treated at another Clinic for Maxillofacial Surgery for extraction of impacted wisdom tooth. A few days after the removal of the threads during feeding, a strong cracking noise was heard in the region of the jaw angle at the side of the extraction. An orthopantomogram and computed tomography were performed, which revealed a pathological fracture. For more information click here.
The editorial in Rare Diseases and Orphan Drugs discussed the main challenges to the long-term sustainability and effectiveness of the European Reference Networks (ERMs), mainly clarifying the legal status of these structures, defining mechanisms of their engagement, collaboration and communication within the national health systems, seeking sources of funding and last but not least, generation of a sustainable product. Now, 2 years later, we come back to this topic. Not all of the above mentioned issues have been resolved, but one thing is clear – the ERMs are here to stay. The second stage of their expansion with new expert centers is expected to start in the autumn of 2020. The current pandemic clearly shows that developing and promoting services in an electronic environment is a high-return investment. The ERMs faces the unique opportunity not only to justify the work and efforts of doctors, patients, health authorities and the academy so far, but also to make a leap into the future. This task is more than ambitious and the challenges are not insignifican – the long-term sustainability and efficiency not only of the ERMs, but of the health systems in the European Union. For more information click here.
The new issue of our scientific journal Rare Diseases and Orphan Drugs is now online. The issue contains 5 publications on various topics. The editorial is “ The European Reference Networks during a pandemic – a step into the future?”. If you would like to receive the latest information on rare diseases and orphan drugs, how the diagnostics and treatment is performed Bulgaria, please follow the link.
 Advancement in technology has improved recognition of genetic etiologies of disease, which has impacted diagnosis and management of rare disease patients in the pediatric pulmonary clinic. This review provides an overview of genetic conditions that are likely to present with pulmonary features and require extensive care by the pediatric pulmonologist. Increased familiarity with these conditions allows for improved care of these patients by reducing time to diagnosis, tailoring management, and prompting further investigation into these disorders. For more information click here.
Advancement in technology has improved recognition of genetic etiologies of disease, which has impacted diagnosis and management of rare disease patients in the pediatric pulmonary clinic. This review provides an overview of genetic conditions that are likely to present with pulmonary features and require extensive care by the pediatric pulmonologist. Increased familiarity with these conditions allows for improved care of these patients by reducing time to diagnosis, tailoring management, and prompting further investigation into these disorders. For more information click here.
 Langerhans cell histiocytosis (LCH) is a clonally expanding neoplasm characterized by the accumulation of CD1a + CD207 + myeloid dendritic cells. As LCH is a rare disease and is presumed to mainly affect children, the clinical features and treatment outcomes of adult LCH have been poorly documented. We retrospectively reviewed 53 adult patients with LCH who were referred to the Institute of Medical Science, the University of Tokyo from 2005 to 2018. The median age at diagnosis was 42 years with a slight female predominance (57%). The time between onset and diagnosis varied among patients (median, 8 months; range, 0-144 months). In total, 40% of the patients had single organ involvement and 60% had multiple organ involvement. Overall, the most frequently affected organ was bone (62%), followed by the central nervous system (34%), and the lung (28%). Twenty-six patients required systemic treatment, and 25 patients underwent the Special C regimen. For more information click here.
Langerhans cell histiocytosis (LCH) is a clonally expanding neoplasm characterized by the accumulation of CD1a + CD207 + myeloid dendritic cells. As LCH is a rare disease and is presumed to mainly affect children, the clinical features and treatment outcomes of adult LCH have been poorly documented. We retrospectively reviewed 53 adult patients with LCH who were referred to the Institute of Medical Science, the University of Tokyo from 2005 to 2018. The median age at diagnosis was 42 years with a slight female predominance (57%). The time between onset and diagnosis varied among patients (median, 8 months; range, 0-144 months). In total, 40% of the patients had single organ involvement and 60% had multiple organ involvement. Overall, the most frequently affected organ was bone (62%), followed by the central nervous system (34%), and the lung (28%). Twenty-six patients required systemic treatment, and 25 patients underwent the Special C regimen. For more information click here.
 Neurofibromatosis type 1 (NF1) syndrome is a common rare/orphan disease that manifests itself early in the paediatric age. It imposes a considerable burden upon patients as well as on caregivers. Decisions regarding optimal care often rely on several medical instances working together as a team. The authors reviewed the literature and supplied a description of their own clinical work at the NF1 centres. The experience of a multidisciplinary teamwork of three NF centres was summarized in order to enhance awareness for possible multidisciplinary ways of delivery of health and health-related aspects of care to NF1 patients. Both population-focused research centres and family-focused centres were reviewed. For more information click here.
Neurofibromatosis type 1 (NF1) syndrome is a common rare/orphan disease that manifests itself early in the paediatric age. It imposes a considerable burden upon patients as well as on caregivers. Decisions regarding optimal care often rely on several medical instances working together as a team. The authors reviewed the literature and supplied a description of their own clinical work at the NF1 centres. The experience of a multidisciplinary teamwork of three NF centres was summarized in order to enhance awareness for possible multidisciplinary ways of delivery of health and health-related aspects of care to NF1 patients. Both population-focused research centres and family-focused centres were reviewed. For more information click here.
 Osteogenesis imperfecta, fibrous dysplasia/McCune-Albright syndrome and X-linked hypophosphatemia are three rare musculoskeletal diseases characterised by bone deformities, frequent fractures and pain. Little high-quality research exists on appropriate treatment and long-term management of these conditions in adults. This is further worsened by limited research funding in rare diseases and a general mismatch between the existing research priorities and those of the patients. This partnership adopted the James Lind Alliance approach to identify the top 10 research priorities for rare musculoskeletal diseases in adults through joint patient, carer and healthcare professional collaboration. For more information click here.
Osteogenesis imperfecta, fibrous dysplasia/McCune-Albright syndrome and X-linked hypophosphatemia are three rare musculoskeletal diseases characterised by bone deformities, frequent fractures and pain. Little high-quality research exists on appropriate treatment and long-term management of these conditions in adults. This is further worsened by limited research funding in rare diseases and a general mismatch between the existing research priorities and those of the patients. This partnership adopted the James Lind Alliance approach to identify the top 10 research priorities for rare musculoskeletal diseases in adults through joint patient, carer and healthcare professional collaboration. For more information click here.
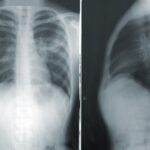 Pulmonary alveolar proteinosis (PAP) is a heterogeneous group of rare diseases characterized by the abnormal production and impaired degradation of pulmonary surfactant as a result of malfunctioning of alveolar macrophages. This is due to the downstream dysregulation of the GM-CSF pathway, which can be caused by specific autoantibodies (autoimmune, aPAP formerly known as idiopathic iPAP), direct injury to alveolar macrophages (e.g. by toxic inhaled agents.), or by genetic defects (hereditary or congenital PAP). Few pharmacotherapy options are currently available to treat this disease. For more information click here.
Pulmonary alveolar proteinosis (PAP) is a heterogeneous group of rare diseases characterized by the abnormal production and impaired degradation of pulmonary surfactant as a result of malfunctioning of alveolar macrophages. This is due to the downstream dysregulation of the GM-CSF pathway, which can be caused by specific autoantibodies (autoimmune, aPAP formerly known as idiopathic iPAP), direct injury to alveolar macrophages (e.g. by toxic inhaled agents.), or by genetic defects (hereditary or congenital PAP). Few pharmacotherapy options are currently available to treat this disease. For more information click here.
 The 11th National Conference for Rare Diseases and Orphan Drugs will be held online on 11-12 September 2020 at the Virtual Congress Venue of the Institute for Rare Diseases. The main topic of the event will be the novelties and current trends in the diagnosis, treatment and follow-up of rare diseases, the development of European reference networks and access to innovation in the field of rare diseases.
The 11th National Conference for Rare Diseases and Orphan Drugs will be held online on 11-12 September 2020 at the Virtual Congress Venue of the Institute for Rare Diseases. The main topic of the event will be the novelties and current trends in the diagnosis, treatment and follow-up of rare diseases, the development of European reference networks and access to innovation in the field of rare diseases.
The registration for medical specialists, PhD students, students, patients and patient organizations is FREE OF CHARGE until 15 August 2020!
The Virtual Congress Venue of the Institute for Rare Diseases is now open for registrations. The website of the Virtual Congress Venue is: https://vcv.raredis.org/.
Subscribe to our YouTube channel, where you can find a short video, explaining how the Internet platform of the Virtual Congress Venue works: https://youtu.be/60ugYXsuQjs
We are expecting you at the Virtual Congress Venue!