
![]() Fabry disease and Pompe disease are rare lysosomal storage disorders that belong to a heterogeneous group of more than 200 distinct inborn metabolic diseases. Mutations followed by loss of function of enzymes or transporters that are localised in the acidic environment of the lysosome may result in degradation of many substrates, such as glycosaminoglycans, glycosphingolipids, glycogen, cholesterol, oligosaccharides, glycoproteins, and peptides, or the excretion of the products degraded by the lysosome. Our aim was to identify the oral signs and symptoms of Fabry disease and Pompe disease from a systematic review made using MEDLINE/PubMed, and a hand search for relevant articles, following the PRISMA guidelines. Both diseases show various craniofacial and oral changes, including supernumerary teeth, dental agenesis, angiokeratoma, and telangiectases in Fabry disease; and macroglossia, teeth fusion, and taurodontism in Pompe disease. Common clinical signs of Fabry disease include hyposalivation, hypohidrosis, and xerophthalmia, and a generally reduced physical resilience was apparent in patients with Pompe disease. Oral and craniofacial changes in patients with both diseases extend over their entire lifetime and can be detected even in an infant. Lysosomal storage diseases should be taken into consideration in the differential diagnosis of relevant diverse symptoms, because treatment, when available, is most effective when started early. The main therapeutic concepts are enzymatic replacement for Pompe disease, whereas patients with Fabry disease require additional oral chaperone treatment or enzyme replacement. For more information click here.
Fabry disease and Pompe disease are rare lysosomal storage disorders that belong to a heterogeneous group of more than 200 distinct inborn metabolic diseases. Mutations followed by loss of function of enzymes or transporters that are localised in the acidic environment of the lysosome may result in degradation of many substrates, such as glycosaminoglycans, glycosphingolipids, glycogen, cholesterol, oligosaccharides, glycoproteins, and peptides, or the excretion of the products degraded by the lysosome. Our aim was to identify the oral signs and symptoms of Fabry disease and Pompe disease from a systematic review made using MEDLINE/PubMed, and a hand search for relevant articles, following the PRISMA guidelines. Both diseases show various craniofacial and oral changes, including supernumerary teeth, dental agenesis, angiokeratoma, and telangiectases in Fabry disease; and macroglossia, teeth fusion, and taurodontism in Pompe disease. Common clinical signs of Fabry disease include hyposalivation, hypohidrosis, and xerophthalmia, and a generally reduced physical resilience was apparent in patients with Pompe disease. Oral and craniofacial changes in patients with both diseases extend over their entire lifetime and can be detected even in an infant. Lysosomal storage diseases should be taken into consideration in the differential diagnosis of relevant diverse symptoms, because treatment, when available, is most effective when started early. The main therapeutic concepts are enzymatic replacement for Pompe disease, whereas patients with Fabry disease require additional oral chaperone treatment or enzyme replacement. For more information click here.
 This guideline was designed to provide service providers and users with an evidence-based set of current best practice guidelines for people and their families and carers, living with epidermolysis bullosa (EB). A systematic literature review relating to the podiatric care of patients with EB was undertaken. Search terms were used, for which the most recent articles relating to podiatric treatment were identified from as early as 1979 to the present day, across seven electronic search engines: MEDLINE, Wiley Online Library, Google Scholar, Athens, ResearchGate, Net and PubFacts.com. The Scottish Intercollegiate Guidelines Network (SIGN) methodology was used. The first guideline draft was analysed and discussed by clinical experts, methodologists and patients and their representatives at four panel meetings. The resulting document went through an external review process by a panel of experts, other healthcare professionals, patient representatives and lay reviewers. The final document will be piloted in three different centres in the U.K. and Australia. Following an EB community international survey the outcomes indicated six main areas that the community indicated as a priority to foot management. These include blistering and wound management, exploring the most suitable footwear and hosiery for EB, management of dystrophic nails, hyperkeratosis (callus), maintaining mobility and fusion of toes (pseudosyndactyly). The evidence here is limited but several interventions currently practised by podiatrists show positive outcomes. For more information click here.
This guideline was designed to provide service providers and users with an evidence-based set of current best practice guidelines for people and their families and carers, living with epidermolysis bullosa (EB). A systematic literature review relating to the podiatric care of patients with EB was undertaken. Search terms were used, for which the most recent articles relating to podiatric treatment were identified from as early as 1979 to the present day, across seven electronic search engines: MEDLINE, Wiley Online Library, Google Scholar, Athens, ResearchGate, Net and PubFacts.com. The Scottish Intercollegiate Guidelines Network (SIGN) methodology was used. The first guideline draft was analysed and discussed by clinical experts, methodologists and patients and their representatives at four panel meetings. The resulting document went through an external review process by a panel of experts, other healthcare professionals, patient representatives and lay reviewers. The final document will be piloted in three different centres in the U.K. and Australia. Following an EB community international survey the outcomes indicated six main areas that the community indicated as a priority to foot management. These include blistering and wound management, exploring the most suitable footwear and hosiery for EB, management of dystrophic nails, hyperkeratosis (callus), maintaining mobility and fusion of toes (pseudosyndactyly). The evidence here is limited but several interventions currently practised by podiatrists show positive outcomes. For more information click here.
 Achondroplasia is the most common form of disproportionate short stature and might affect not only the quality of life of the affected child but also that of the parents. The aim is to investigate the quality of life of children with achondroplasia from child- and parent perspective as well as the parental quality of life. Forty-seven children with achondroplasia and 73 parents from a German patient organization participated. We assessed children’s quality of life using the generic Peds QL 4.0™ as self-reports for children aged 8-14 and parent-reports for children aged 4-14 years. Parental quality of life we assessed using the short-form 8-questionnaire. Achondroplasia is chronically debilitating. Thus special efforts are needed to address patients’ and parent’s quality of life needs. This special health condition may influence the daily life of the entire family because they have to adapt to the child’s particular needs. Therefore, clinicians should not only focus on the child’s quality of life but also those of the parents. For more information click here.
Achondroplasia is the most common form of disproportionate short stature and might affect not only the quality of life of the affected child but also that of the parents. The aim is to investigate the quality of life of children with achondroplasia from child- and parent perspective as well as the parental quality of life. Forty-seven children with achondroplasia and 73 parents from a German patient organization participated. We assessed children’s quality of life using the generic Peds QL 4.0™ as self-reports for children aged 8-14 and parent-reports for children aged 4-14 years. Parental quality of life we assessed using the short-form 8-questionnaire. Achondroplasia is chronically debilitating. Thus special efforts are needed to address patients’ and parent’s quality of life needs. This special health condition may influence the daily life of the entire family because they have to adapt to the child’s particular needs. Therefore, clinicians should not only focus on the child’s quality of life but also those of the parents. For more information click here.
 Limited data exist about the clinical presentation, ideal therapy and outcomes of patients with hereditary hemorrhagic telangiectasia (HHT) who develop venous thromboembolism (VTE). We used the data in the RIETE Registry to assess the clinical characteristics, therapeutic approaches and clinical outcomes during the course of anticoagulant therapy in patients with HHT according to initial presentation as pulmonary embolism (PE) or deep venous thrombosis (DVT). Of 51,375 patients with acute VTE enrolled in RIETE from February 2009 to January 2019, 23 (0.04%) had HHT: 14 (61%) initially presented with PE and 9 (39%) with DVT alone. Almost half (47.8%) of the patients with VTE had a risk factor for VTE. Most PE and DVT patients received low-molecular-weight heparin for initial (71 and 100%, respectively) and long-term therapy (54 and 67%, respectively). During anticoagulation for VTE, the rate of bleeding events (major 2, non-major 6) far outweighed the rate of VTE recurrences (recurrent DVT 1): 50.1 bleeds per 100 patient-years (95%CI: 21.6-98.7) vs. 6.26 recurrences (95%CI: 0.31-30.9; p = 0.020). One major and three non-major bleeding were epistaxis. No patient died of bleeding. One patient died shortly after being diagnosed with acute PE. During anticoagulation for VTE in HHT patients, there were more bleeding events than VTE recurrences. Most bleeding episodes were non-major epistaxis. For more information click here.
Limited data exist about the clinical presentation, ideal therapy and outcomes of patients with hereditary hemorrhagic telangiectasia (HHT) who develop venous thromboembolism (VTE). We used the data in the RIETE Registry to assess the clinical characteristics, therapeutic approaches and clinical outcomes during the course of anticoagulant therapy in patients with HHT according to initial presentation as pulmonary embolism (PE) or deep venous thrombosis (DVT). Of 51,375 patients with acute VTE enrolled in RIETE from February 2009 to January 2019, 23 (0.04%) had HHT: 14 (61%) initially presented with PE and 9 (39%) with DVT alone. Almost half (47.8%) of the patients with VTE had a risk factor for VTE. Most PE and DVT patients received low-molecular-weight heparin for initial (71 and 100%, respectively) and long-term therapy (54 and 67%, respectively). During anticoagulation for VTE, the rate of bleeding events (major 2, non-major 6) far outweighed the rate of VTE recurrences (recurrent DVT 1): 50.1 bleeds per 100 patient-years (95%CI: 21.6-98.7) vs. 6.26 recurrences (95%CI: 0.31-30.9; p = 0.020). One major and three non-major bleeding were epistaxis. No patient died of bleeding. One patient died shortly after being diagnosed with acute PE. During anticoagulation for VTE in HHT patients, there were more bleeding events than VTE recurrences. Most bleeding episodes were non-major epistaxis. For more information click here.
 This year on 6-8 of December 2019 in Athens, Greece the Duchenne Data Foundation, with the support of the World Duchenne Organization, is launching the 3rd edition of the Duchenne Patient Academy. Over the past years, the Duchenne world has been changing due to a fast evolving research and regulatory landscape. For this, the need for adaptive and proactive Duchenne advocates has increased. In this intensive two-day training session, both Next Generation Young Advocates and Experienced Advocates will receive a parallel training and updates to set a strong base for current and future global advocacy.This Duchenne Masterclass will focus on setting the foundation of advocacy and gaining a deeper understanding of the Duchenne field in terms of regulatory, HTA, clinical trial, care, research and industry developments. For more information click here.
This year on 6-8 of December 2019 in Athens, Greece the Duchenne Data Foundation, with the support of the World Duchenne Organization, is launching the 3rd edition of the Duchenne Patient Academy. Over the past years, the Duchenne world has been changing due to a fast evolving research and regulatory landscape. For this, the need for adaptive and proactive Duchenne advocates has increased. In this intensive two-day training session, both Next Generation Young Advocates and Experienced Advocates will receive a parallel training and updates to set a strong base for current and future global advocacy.This Duchenne Masterclass will focus on setting the foundation of advocacy and gaining a deeper understanding of the Duchenne field in terms of regulatory, HTA, clinical trial, care, research and industry developments. For more information click here.
 NCATS is dedicated to engaging the patient community throughout the translational science process. The NCATS Toolkit for Patient-Focused Therapy Development was created to provide a collection of online resources that can help patient groups advance through the process of therapy development and provide them with the tools they need to advance medical research. Launched in September 2017, the Toolkit includes resources that have been developed primarily for the rare diseases community to facilitate therapeutics research and development. Since early 2016, NCATS has worked with a diverse group of partners in the rare diseases community to conduct an extensive landscape analysis of available tools. These resources were defined, characterized and organized in a centralized portal that can be helpful to all patient groups regardless of how far along in the research and development process they might be. For more information click here.
NCATS is dedicated to engaging the patient community throughout the translational science process. The NCATS Toolkit for Patient-Focused Therapy Development was created to provide a collection of online resources that can help patient groups advance through the process of therapy development and provide them with the tools they need to advance medical research. Launched in September 2017, the Toolkit includes resources that have been developed primarily for the rare diseases community to facilitate therapeutics research and development. Since early 2016, NCATS has worked with a diverse group of partners in the rare diseases community to conduct an extensive landscape analysis of available tools. These resources were defined, characterized and organized in a centralized portal that can be helpful to all patient groups regardless of how far along in the research and development process they might be. For more information click here.
 Colorectal neuroendocrine carcinoma (NEC) is a rare disease, and mixed cases with colorectal adenocarcinoma also exist. The histogenesis of this disease remains unclear. We studied the numbers of neuroendocrine marker-positive cells in adenocarcinoma tissue and in normal -mucosal tissue to investigate the relation between adenocarcinoma and NEC and to discuss the histogenesis of NEC. Among the 390 cases, 181 cases had right sided colon cancer, 173 cases had left sided colon cancer, and 36 cases had rectal cancer. The rates of positive staining for chromogranin A, synaptophysin, and CD56 were significantly higher in the right sided colon than in the left sided colon, consistent with the preferred sites of NEC as reported previously. Cells positive for chromogranin A and synaptophysin in normal mucosa were significantly more common in the rectum and the left sided colon than in the right sided colon. No site-specific differences were found for CD56. Neuroendocrine marker-positive cells in colorectal cancer tissue are more common in the right sided colon, whereas neuroendocrine marker-positive cells in normal mucosa are more common in the rectum. These results suggest that NEC may arise from preceding adenocarcinomas. For more information click here.
Colorectal neuroendocrine carcinoma (NEC) is a rare disease, and mixed cases with colorectal adenocarcinoma also exist. The histogenesis of this disease remains unclear. We studied the numbers of neuroendocrine marker-positive cells in adenocarcinoma tissue and in normal -mucosal tissue to investigate the relation between adenocarcinoma and NEC and to discuss the histogenesis of NEC. Among the 390 cases, 181 cases had right sided colon cancer, 173 cases had left sided colon cancer, and 36 cases had rectal cancer. The rates of positive staining for chromogranin A, synaptophysin, and CD56 were significantly higher in the right sided colon than in the left sided colon, consistent with the preferred sites of NEC as reported previously. Cells positive for chromogranin A and synaptophysin in normal mucosa were significantly more common in the rectum and the left sided colon than in the right sided colon. No site-specific differences were found for CD56. Neuroendocrine marker-positive cells in colorectal cancer tissue are more common in the right sided colon, whereas neuroendocrine marker-positive cells in normal mucosa are more common in the rectum. These results suggest that NEC may arise from preceding adenocarcinomas. For more information click here.
 Systemic sclerosis (SSc) is a rare disease characterized by widespread collagen deposition resulting in fibrosis. Although skin involvement is the most common manifestation and also the one that determines the classification of disease, mortality in SSc is usually a result of respiratory compromise in the form of interstitial lung disease (ILD) or pulmonary hypertension (PH). Clinically significant ILD is seen in up to 40% of patients and PH in up to 20%. Treatment with either cyclophosphamide or mycophenolate has been shown to delay disease progression, whereas rituximab and lung transplantation are reserved for refractory cases. For more information click here.
Systemic sclerosis (SSc) is a rare disease characterized by widespread collagen deposition resulting in fibrosis. Although skin involvement is the most common manifestation and also the one that determines the classification of disease, mortality in SSc is usually a result of respiratory compromise in the form of interstitial lung disease (ILD) or pulmonary hypertension (PH). Clinically significant ILD is seen in up to 40% of patients and PH in up to 20%. Treatment with either cyclophosphamide or mycophenolate has been shown to delay disease progression, whereas rituximab and lung transplantation are reserved for refractory cases. For more information click here.
 The First National Congress on Tarlov Perineural Cysts will be held in Stara Zagora on November 9 and 10, 2019 and it will be attended by leading specialists in the fields of neurology, neurosurgery, kinesitherapy and psychology. Patient organizations will also be part of the forum. The purpose of the event is to promote the disease, to inform and help people and also to explain to the patients the treatment options and their rights. For more information click here.
The First National Congress on Tarlov Perineural Cysts will be held in Stara Zagora on November 9 and 10, 2019 and it will be attended by leading specialists in the fields of neurology, neurosurgery, kinesitherapy and psychology. Patient organizations will also be part of the forum. The purpose of the event is to promote the disease, to inform and help people and also to explain to the patients the treatment options and their rights. For more information click here.
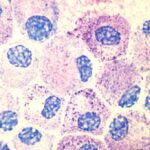 Systemic Mastocytosis (SM) is a complex family of rare diseases, against which pharmacological therapies are still very few. It is a c-kit driven disease, whose disregulation leads to uncontrolled activation and proliferation of mast cells (MCs) with consequent release of effector molecules which are responsible for its clinical manifestations. Masitinib is a relatively new potential drug against SM and its chemical structure strictly derives from imatinib, the first tyrosine kinase inhibitor which entered the pharmaceutical market about 15 years ago. In this review, the authors present masitinib in all its properties, from chemistry to pharmacology and toxicity to its potential clinical application in SM, focusing the discussion on the few clinical trials in which it has been involved, with a particular attention on the still open challenge to determine how to measure the response to therapy. In spite of their similarity in chemistry and biological activity against submolecular targets, masitinib is much more selective towards c-kit receptors than other tyrosine kinases, such as Bcl-Abl. Furthermore, its ability to inhibit degranulation, cytokine production and MCs migration from bone marrow gives it a great chance to become an important therapeutic option for selected SM patients. For more information click here.
Systemic Mastocytosis (SM) is a complex family of rare diseases, against which pharmacological therapies are still very few. It is a c-kit driven disease, whose disregulation leads to uncontrolled activation and proliferation of mast cells (MCs) with consequent release of effector molecules which are responsible for its clinical manifestations. Masitinib is a relatively new potential drug against SM and its chemical structure strictly derives from imatinib, the first tyrosine kinase inhibitor which entered the pharmaceutical market about 15 years ago. In this review, the authors present masitinib in all its properties, from chemistry to pharmacology and toxicity to its potential clinical application in SM, focusing the discussion on the few clinical trials in which it has been involved, with a particular attention on the still open challenge to determine how to measure the response to therapy. In spite of their similarity in chemistry and biological activity against submolecular targets, masitinib is much more selective towards c-kit receptors than other tyrosine kinases, such as Bcl-Abl. Furthermore, its ability to inhibit degranulation, cytokine production and MCs migration from bone marrow gives it a great chance to become an important therapeutic option for selected SM patients. For more information click here.