
 Саркоидозата (SA) не е забооляване само на белите дробове, но може да засегне и други органи, като черния дроб и далака и поради тази причина се полагат усилия за определяне на специфични критерии за изобразяване и диагностициране на всеки един орган, като основната концепция е изключването на други причини за заболяването и е важно за постигане на правилната диагноза. Ултразвукът (US) е полезен инструмент за оценка на пациенти със съмнение за коремна SA, като например при засягане на черния дроб, далака, бъбреците, панкреаса и други органи, показващи находки като органомегалия, фокални лезии и лимфаденопатия. Докато диагностицирането на коремната SA е по-предсказуемо в случай на включване на други органи (например, бели дробове), проблемът е по-сложен в случая на изолирана коремна SA. Употребата на ултразвук и ендоскопска ултразвукова еластография с контрастно усилване предостави допълнителна информация за моделите на усилване и плътността на тъканите при коремната SA. Повече информация може да получите тук.
Саркоидозата (SA) не е забооляване само на белите дробове, но може да засегне и други органи, като черния дроб и далака и поради тази причина се полагат усилия за определяне на специфични критерии за изобразяване и диагностициране на всеки един орган, като основната концепция е изключването на други причини за заболяването и е важно за постигане на правилната диагноза. Ултразвукът (US) е полезен инструмент за оценка на пациенти със съмнение за коремна SA, като например при засягане на черния дроб, далака, бъбреците, панкреаса и други органи, показващи находки като органомегалия, фокални лезии и лимфаденопатия. Докато диагностицирането на коремната SA е по-предсказуемо в случай на включване на други органи (например, бели дробове), проблемът е по-сложен в случая на изолирана коремна SA. Употребата на ултразвук и ендоскопска ултразвукова еластография с контрастно усилване предостави допълнителна информация за моделите на усилване и плътността на тъканите при коремната SA. Повече информация може да получите тук.
 Синдромът на Сатайоши е мултисистемно рядко заболяване с неизвестна етиология, въпреки че се предполага автоимунна основа. Неговите основни симптоми са: болезнени мускулни спазми, диария, алопеция и скелетни аномалии. Клиничният курс без лечение може да доведе до сериозно увреждане или смърт. Все още предстои преглед на лечението и неговия отговор. Между 1967 и 2018 г. са публикувани 64 случая на синдром на Сатайоши. Използваните лекарства могат да бъдат разделени на две основни групи на лечение: мускулни релаксанти / антиконвулсанти и кортикостероиди / имуносупресори. Дантролен подобрява мускулните симптоми при 13 от 15 случая, но не и други симптоми на заболяването. Други мускулни релаксанти или антиконвулсивни лекарствени средства показват малък или никакъв ефект. 28 от 30 случая отговарят на схема, включваща костикостероиди. Други имуносупресивни лекарства, включително циклоспорин, микофенолат мофетил, азатиоприн, метотрексат, такролимус и циклофосфамид са използвани с цел намаляване на дозата на кортикостероидите или за подобряване на ефикасността. Имуноглобулинова терапия е използвана при девет пациента и четири от тях са получили благоприятен отговор. Кортикостероидите са най-широко използваното лечение с най-добри резултати при синдрома на Сатайоши. Необходими са допълнителни проучвания, за да се определи оптималната доза и продължителността на кортикостероидния курс, както и ролята на други имуносупресори и имуноглобулинова терапия. Генетични или автоимунни маркери ще бъдат полезни за насочване на бъдещите терапии. Повече информация може да получите тук.
Синдромът на Сатайоши е мултисистемно рядко заболяване с неизвестна етиология, въпреки че се предполага автоимунна основа. Неговите основни симптоми са: болезнени мускулни спазми, диария, алопеция и скелетни аномалии. Клиничният курс без лечение може да доведе до сериозно увреждане или смърт. Все още предстои преглед на лечението и неговия отговор. Между 1967 и 2018 г. са публикувани 64 случая на синдром на Сатайоши. Използваните лекарства могат да бъдат разделени на две основни групи на лечение: мускулни релаксанти / антиконвулсанти и кортикостероиди / имуносупресори. Дантролен подобрява мускулните симптоми при 13 от 15 случая, но не и други симптоми на заболяването. Други мускулни релаксанти или антиконвулсивни лекарствени средства показват малък или никакъв ефект. 28 от 30 случая отговарят на схема, включваща костикостероиди. Други имуносупресивни лекарства, включително циклоспорин, микофенолат мофетил, азатиоприн, метотрексат, такролимус и циклофосфамид са използвани с цел намаляване на дозата на кортикостероидите или за подобряване на ефикасността. Имуноглобулинова терапия е използвана при девет пациента и четири от тях са получили благоприятен отговор. Кортикостероидите са най-широко използваното лечение с най-добри резултати при синдрома на Сатайоши. Необходими са допълнителни проучвания, за да се определи оптималната доза и продължителността на кортикостероидния курс, както и ролята на други имуносупресори и имуноглобулинова терапия. Генетични или автоимунни маркери ще бъдат полезни за насочване на бъдещите терапии. Повече информация може да получите тук.
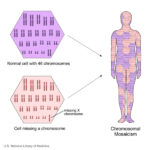 Синдромът на Търнър е рядко състояние при жените, което е свързано или с пълна, или с частична загуба на една Х-хромозома, често в мозаечни кариотипове. Синдромът на Търнър се свързва с нисък ръст, забавен пубертет, яйчникова дисгенезия, хипергонадотропен хипогонадизъм, безплодие, вродени малформации на сърцето, ендокринни заболявания като захарен диабет тип 1 и тип 2, остеопороза и автоимунни заболявания. Заболеваемостта и смъртността са по-големи при жени със синдром на Търнър в сравнение с общото население и засягането на множество органи и системи през всички етапи от живота изисква мултидисциплинарен подход в лечението. Въпреки характерния фенотип, диагностичното забавяне може да бъде значително и средната възраст за поставяне на диагнозата е около 15-годишна възраст. Въпреки това, са постигнати много важни клинични постижения, обхващащи всички специалности, включени в грижата за момичета и жени със синдром на Търнър. Статията представя актуализиран преглед на синдрома на Търнър, който обхваща напредъка в генетичните и геномни механизми на заболяването, свързаните с него нарушения и мултидисциплинарните подходи в лечението на пациентите, включително и терапия с растежен хормон и хормонозаместителна терапия. Повече информация може да получите тук.
Синдромът на Търнър е рядко състояние при жените, което е свързано или с пълна, или с частична загуба на една Х-хромозома, често в мозаечни кариотипове. Синдромът на Търнър се свързва с нисък ръст, забавен пубертет, яйчникова дисгенезия, хипергонадотропен хипогонадизъм, безплодие, вродени малформации на сърцето, ендокринни заболявания като захарен диабет тип 1 и тип 2, остеопороза и автоимунни заболявания. Заболеваемостта и смъртността са по-големи при жени със синдром на Търнър в сравнение с общото население и засягането на множество органи и системи през всички етапи от живота изисква мултидисциплинарен подход в лечението. Въпреки характерния фенотип, диагностичното забавяне може да бъде значително и средната възраст за поставяне на диагнозата е около 15-годишна възраст. Въпреки това, са постигнати много важни клинични постижения, обхващащи всички специалности, включени в грижата за момичета и жени със синдром на Търнър. Статията представя актуализиран преглед на синдрома на Търнър, който обхваща напредъка в генетичните и геномни механизми на заболяването, свързаните с него нарушения и мултидисциплинарните подходи в лечението на пациентите, включително и терапия с растежен хормон и хормонозаместителна терапия. Повече информация може да получите тук.
 Анемията на Diamond-Blackfan е рядка вродена аплазия на червените кръвни клетки, характеризираща се с неуспешна еритропоеза, вродени аномалии в до 50% от пациентите, забавяне на растежа в до 30% от пациентите и предразположеност към злокачествени заболявания. Заболяването е както клинично, така и генетично хетерогенно състояние, вариращо от фини асимптоматични еритроидни аномалии до неимунен фетален хидропс. Съвременните възможности за лечение включват кортикостероидна терапия, хронични трансфузии на червени кръвни клетки и трансплантация на хемопоетични стволови клетки с генна терапия. В публикацията се съобщава за първия документиран случай на анемията на Diamond-Blackfan в Южна Африка при едно европеидно момиче с вторична хетерозиготна делеция на ген в RPL35A. Ограничените ресурси, липсата на тестове, непознаването на редките заболявания, разширената диференциална диагноза и свързаната неутропения са довели до забавяне в диагностиката на заболяването. Този случай напомня на клиничните специалисти, че това заболяване може да доведе и до апластична анемия и подчертава трудностите и пречките при диагностицирането му в ограничените от ресурси страни. Случаят напомня още, че може да е причина за анемия при бебетата, за ограниченията при поставянето на диагноза в здравните системи с недостатъчни ресурси и необходимостта от стандартизирани протоколи за лечение, приложими за страните с ограничен ресурс. Повече информация може да получите тук.
Анемията на Diamond-Blackfan е рядка вродена аплазия на червените кръвни клетки, характеризираща се с неуспешна еритропоеза, вродени аномалии в до 50% от пациентите, забавяне на растежа в до 30% от пациентите и предразположеност към злокачествени заболявания. Заболяването е както клинично, така и генетично хетерогенно състояние, вариращо от фини асимптоматични еритроидни аномалии до неимунен фетален хидропс. Съвременните възможности за лечение включват кортикостероидна терапия, хронични трансфузии на червени кръвни клетки и трансплантация на хемопоетични стволови клетки с генна терапия. В публикацията се съобщава за първия документиран случай на анемията на Diamond-Blackfan в Южна Африка при едно европеидно момиче с вторична хетерозиготна делеция на ген в RPL35A. Ограничените ресурси, липсата на тестове, непознаването на редките заболявания, разширената диференциална диагноза и свързаната неутропения са довели до забавяне в диагностиката на заболяването. Този случай напомня на клиничните специалисти, че това заболяване може да доведе и до апластична анемия и подчертава трудностите и пречките при диагностицирането му в ограничените от ресурси страни. Случаят напомня още, че може да е причина за анемия при бебетата, за ограниченията при поставянето на диагноза в здравните системи с недостатъчни ресурси и необходимостта от стандартизирани протоколи за лечение, приложими за страните с ограничен ресурс. Повече информация може да получите тук.
 Хемофилия А (НА) е Х-свързано наследствено нарушение на кръвта, причинено от дефицит на коагулационен фактор VIII (FVIII). Едно от най-големите усложнения при лечението на НА е развитието на неутрализиращи алоантитела, известни като инхибитори на FVIII. Пациентите, страдащи от HA, при които се образуват инхибитори на FVIII, имат ограничени възможности за лечение и срещат по-големи проблеми, свързани с лечението и заболяването, отколкото пациентите на HA без FVIII инхибитори. Емицизумаб, наскоро одобрено биспецифично моноклонално антитяло, имитира функцията на FVIIIa чрез свързване на FIXa и FX за възстановяване на ефективната хемостаза. В публикацията се обсъждат текущите лабораторни методи за мониторинг, включително активирано парциално тромбопластиново време, FVIII едноетапни анализи на кръвосъсирването, FVIII хромогенни анализи и глобални анализи на коагулациите; посочва се защо тези конвенционални методи могат да бъдат неподходящи за наблюдение на пациенти с НА, получаващи емицизумаб и се предлагат алтернативни методи, приложими за мониториране на лечението на НА в един развиващ се пейзаж. Повече информация може да получите тук.
Хемофилия А (НА) е Х-свързано наследствено нарушение на кръвта, причинено от дефицит на коагулационен фактор VIII (FVIII). Едно от най-големите усложнения при лечението на НА е развитието на неутрализиращи алоантитела, известни като инхибитори на FVIII. Пациентите, страдащи от HA, при които се образуват инхибитори на FVIII, имат ограничени възможности за лечение и срещат по-големи проблеми, свързани с лечението и заболяването, отколкото пациентите на HA без FVIII инхибитори. Емицизумаб, наскоро одобрено биспецифично моноклонално антитяло, имитира функцията на FVIIIa чрез свързване на FIXa и FX за възстановяване на ефективната хемостаза. В публикацията се обсъждат текущите лабораторни методи за мониторинг, включително активирано парциално тромбопластиново време, FVIII едноетапни анализи на кръвосъсирването, FVIII хромогенни анализи и глобални анализи на коагулациите; посочва се защо тези конвенционални методи могат да бъдат неподходящи за наблюдение на пациенти с НА, получаващи емицизумаб и се предлагат алтернативни методи, приложими за мониториране на лечението на НА в един развиващ се пейзаж. Повече информация може да получите тук.
 Редките очни патологии имат важно влияние върху качеството на живот на пациентите, тъй като често увреждането е двустранно и въпреки, че са асиметрични причиняват значително намаляване на зрителната острота. Често могат да протекат асимптоматично до сравнително късен етап и поради това диагнозата често се забавя. Разбирането на патофизиологията, диагнозата и лечението на заболяването може да помогне на лекарите от първичната медицинска помощ да насочат пациентите с висок риск за цялостно офталмологично изследване и за по-активно участие в грижите за себе си. Голям процент от тези редки болести нямат лечение, което да е одобрено от FDA. Изследването и мониторинга на пациенти с редки офталмологични нарушения представлява ключов компонент в проекта на Университетската спешна болница, Букурещ, Румъния – Клиника по офталмология. Регистрите с редки болести са водещи инструменти за разработване на клинични изследвания за редки заболявания, подобряване на достъпа на пациентите до нови диагностични методи, проследяване и нови терапии. Към настоящия момент европейският списък на редките заболявания включва 53 офталмологични заболявания, които са класифицирани като редки болести и други 103 системни заболявания с офталмологично засягане, от общо 7000 редки заболявания. Повече информация може да получите тук.
Редките очни патологии имат важно влияние върху качеството на живот на пациентите, тъй като често увреждането е двустранно и въпреки, че са асиметрични причиняват значително намаляване на зрителната острота. Често могат да протекат асимптоматично до сравнително късен етап и поради това диагнозата често се забавя. Разбирането на патофизиологията, диагнозата и лечението на заболяването може да помогне на лекарите от първичната медицинска помощ да насочат пациентите с висок риск за цялостно офталмологично изследване и за по-активно участие в грижите за себе си. Голям процент от тези редки болести нямат лечение, което да е одобрено от FDA. Изследването и мониторинга на пациенти с редки офталмологични нарушения представлява ключов компонент в проекта на Университетската спешна болница, Букурещ, Румъния – Клиника по офталмология. Регистрите с редки болести са водещи инструменти за разработване на клинични изследвания за редки заболявания, подобряване на достъпа на пациентите до нови диагностични методи, проследяване и нови терапии. Към настоящия момент европейският списък на редките заболявания включва 53 офталмологични заболявания, които са класифицирани като редки болести и други 103 системни заболявания с офталмологично засягане, от общо 7000 редки заболявания. Повече информация може да получите тук.
 За десета поредна година Института за редки болести и лекарства сираци организира национална конференция. И тази година конференцията ще се проведе под официалния патронаж на Комисията по здравеопазването към 44-то Народно събрание на Република България.
За десета поредна година Института за редки болести и лекарства сираци организира национална конференция. И тази година конференцията ще се проведе под официалния патронаж на Комисията по здравеопазването към 44-то Народно събрание на Република България.
“В рамките на своите правомощия, Комисията по здравеопазването винаги е била отворен партньор в подкрепа на усилията на институциите и неправителствените организации за подобряване развитието на профилактиката, диагностиката, лечението, рехабилитацията и социалните грижи за хората с редки заболявания“, посочи в писмото си д-р Даниела Дариткова.
Основна тема на събитието ще бъдат новостите и актуалните тенденции в диагностиката, лечението и проследяването на редките болести, развитието на европейските референти мрежи и достъпа до иновации в областта на редки болести. По време на събитието ще бъде отбелязана и 15-годишнината на Информационен център за редки болести и лекарства сираци (ИЦРБЛС). Информационният център стартира своята дейност през 2004 г. като първият информационен център по рода си в Източна Европа, посветен на пациенти, организации и медицински специалисти с интерес към редките болести и лекарствата сираци. ИЦРБЛС се развива през тези 15 години с подкрепата и помощта на 26 доброволни консултанти – водещи медицински специалисти от университетските клиники в България. Повече информация за събитието може да получите тук.
 Постигането на нормален растеж е един от най-трудните проблеми при лечението на деца с хронично бъбречно заболяване (ХБЗ). Лечението с рекомбинантен човешки растежен хормон (РХ) насърчава надлъжния растеж и вероятно дава възможност на децата с ХБЗ и нисък ръст да достигнат нормална височина. Членовете на Европейската група за детска нефрология представят препоръки за клинична практика при използване на РХ при деца с ХБЗ на диализа и след бъбречна трансплантация. Тези препоръки са разработени с помощта на външна консултативна група от детски ендокринолози, нефролози и представители на пациенти. Препоръчва се децата с ХБЗ в 3-5 стадий или на диализа да бъдат кандидати за терапия с РХ ако изостават в ръста си, при условие, че детето има потенциал за растеж. При деца, които са трансплантирани се препоръчва започване на терапия с РХ една година след трансплантацията, само ако не възникне спонтанен догонващ растеж и ако безстероидната имуносупресията не е възможен вариант. РХ трябва да се прилага в дози от 0,045-0,05 mg / kg дневно чрез ежедневни подкожни инжекции, докато пациентът достигне крайната си височина или до осъществяването на бъбречна трансплантация. Повече информация може да получите тук.
Постигането на нормален растеж е един от най-трудните проблеми при лечението на деца с хронично бъбречно заболяване (ХБЗ). Лечението с рекомбинантен човешки растежен хормон (РХ) насърчава надлъжния растеж и вероятно дава възможност на децата с ХБЗ и нисък ръст да достигнат нормална височина. Членовете на Европейската група за детска нефрология представят препоръки за клинична практика при използване на РХ при деца с ХБЗ на диализа и след бъбречна трансплантация. Тези препоръки са разработени с помощта на външна консултативна група от детски ендокринолози, нефролози и представители на пациенти. Препоръчва се децата с ХБЗ в 3-5 стадий или на диализа да бъдат кандидати за терапия с РХ ако изостават в ръста си, при условие, че детето има потенциал за растеж. При деца, които са трансплантирани се препоръчва започване на терапия с РХ една година след трансплантацията, само ако не възникне спонтанен догонващ растеж и ако безстероидната имуносупресията не е възможен вариант. РХ трябва да се прилага в дози от 0,045-0,05 mg / kg дневно чрез ежедневни подкожни инжекции, докато пациентът достигне крайната си височина или до осъществяването на бъбречна трансплантация. Повече информация може да получите тук.
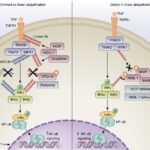 Моногенните автовъзпалителни заболявания са редки заболявания, причинени от гени, регулиращи вродения имунитет. Идентифицирането на първите четири гена, отговорни за прототипа на наследствените рецидиви е предизвикало развитие в генетичната диагностика, последвано от външна оценка на качеството и насоки за тълкуване на различните варианти за секвениране при тези заболявания. Последните промени в диагностиката на генетичните заболявания, а именно прилагането на секвениране от новото поколение (NGS), водят до откриването на 40 нови гена, свързани с автовъзпалителни заболявания, които революционизират грижите и прогнозите на пациентите. Тези бързи постижения обаче са довели до нестандартни молекулярни стратегии, които могат да повлияят на генетичната диагностика и докладването на резултатите. За да се оценят факторите, които могат да окажат въздействие върху резултатите и качеството на резултатите в епохата на NGS е осъществено онлайн проучване сред лабораториите, членки на Европейската мрежа за качество на молекулярната генетика, в която са изтъкнати различни стратегии и са установени проблемите, които е необходимо да се дискутират и подобрят. Повече информация може да получите тук.
Моногенните автовъзпалителни заболявания са редки заболявания, причинени от гени, регулиращи вродения имунитет. Идентифицирането на първите четири гена, отговорни за прототипа на наследствените рецидиви е предизвикало развитие в генетичната диагностика, последвано от външна оценка на качеството и насоки за тълкуване на различните варианти за секвениране при тези заболявания. Последните промени в диагностиката на генетичните заболявания, а именно прилагането на секвениране от новото поколение (NGS), водят до откриването на 40 нови гена, свързани с автовъзпалителни заболявания, които революционизират грижите и прогнозите на пациентите. Тези бързи постижения обаче са довели до нестандартни молекулярни стратегии, които могат да повлияят на генетичната диагностика и докладването на резултатите. За да се оценят факторите, които могат да окажат въздействие върху резултатите и качеството на резултатите в епохата на NGS е осъществено онлайн проучване сред лабораториите, членки на Европейската мрежа за качество на молекулярната генетика, в която са изтъкнати различни стратегии и са установени проблемите, които е необходимо да се дискутират и подобрят. Повече информация може да получите тук.
 Наследственият ангиоедем (HAE) не трябва да се пренебрегва. Освен подуване на кожата, тези пациенти могат да имат много болезнени абдоминални атаки и потенциално животозастрашаващ ангиоедем на горните дихателни пътища. Те не отговорят на традиционната антиалергична терапия с антихистамини, кортикостероиди и адреналин, а вместо това се нуждаят от специфични лекарства, насочени към каликреин-кининовия път. Пациентите с HAE имат количествен или качествен дефицит на С1 инхибитора (C1INH) поради различни мутации в SERPING1, въпреки че в последно време е признат нов подтип с нормален C1INH. Последният вариант се диагностицира въз основа на клинични признаци, фамилна анамнеза или молекулярно-генетично изследване за мутации в F12, ANGPT1 или PLG. Диагнозата HAE често се забавя поради общо незнание за това заболяване. Недиагностицираните пациенти обаче са изложени на повишен риск от ненужни хирургически интервенции или животозастрашаващ ларингеален оток. През последното десетилетие са разработени и въведени нови и ефективни терапии за профилактика и лечение на острите прояви. Още повече лекарства се оценяват в клинични проучвания. Ето защо е изключително важно пациентите с HAE да се диагностицират възможно най-скоро и да се предложи съответна терапия с лекарства сираци за да се намали заболеваемостта, да се предотврати смъртността и да се подобри качеството на живот. Повече информация може да получите тук.
Наследственият ангиоедем (HAE) не трябва да се пренебрегва. Освен подуване на кожата, тези пациенти могат да имат много болезнени абдоминални атаки и потенциално животозастрашаващ ангиоедем на горните дихателни пътища. Те не отговорят на традиционната антиалергична терапия с антихистамини, кортикостероиди и адреналин, а вместо това се нуждаят от специфични лекарства, насочени към каликреин-кининовия път. Пациентите с HAE имат количествен или качествен дефицит на С1 инхибитора (C1INH) поради различни мутации в SERPING1, въпреки че в последно време е признат нов подтип с нормален C1INH. Последният вариант се диагностицира въз основа на клинични признаци, фамилна анамнеза или молекулярно-генетично изследване за мутации в F12, ANGPT1 или PLG. Диагнозата HAE често се забавя поради общо незнание за това заболяване. Недиагностицираните пациенти обаче са изложени на повишен риск от ненужни хирургически интервенции или животозастрашаващ ларингеален оток. През последното десетилетие са разработени и въведени нови и ефективни терапии за профилактика и лечение на острите прояви. Още повече лекарства се оценяват в клинични проучвания. Ето защо е изключително важно пациентите с HAE да се диагностицират възможно най-скоро и да се предложи съответна терапия с лекарства сираци за да се намали заболеваемостта, да се предотврати смъртността и да се подобри качеството на живот. Повече информация може да получите тук.