
 Since it has been recognized that sarcoidosis (SA) is not an exclusive disorder of the lungs but can also affect other organs such as the liver and spleen, efforts have been made to define specific imaging criteria for the diagnosis of the single organ involvement, and the concept has been reinforced that the exclusion of alternative causes is important to achieve the correct diagnosis. Ultrasound (US) is a useful tool to evaluate patients with suspected abdominal SA, such as of the liver, spleen, kidney, pancreas and other organs, showing findings such as organomegaly, focal lesions and lymphadenopathy. While the diagnosis of abdominal SA is more predictable in the case of involvement of other organs (e.g., lungs), the problem is more complex in the case of isolated abdominal SA. The recent use of contrast-enhanced ultrasound and endoscopic ultrasound elastography has provided additional information about the enhancement patterns and tissue rigidity in abdominal SA. For more information click here.
Since it has been recognized that sarcoidosis (SA) is not an exclusive disorder of the lungs but can also affect other organs such as the liver and spleen, efforts have been made to define specific imaging criteria for the diagnosis of the single organ involvement, and the concept has been reinforced that the exclusion of alternative causes is important to achieve the correct diagnosis. Ultrasound (US) is a useful tool to evaluate patients with suspected abdominal SA, such as of the liver, spleen, kidney, pancreas and other organs, showing findings such as organomegaly, focal lesions and lymphadenopathy. While the diagnosis of abdominal SA is more predictable in the case of involvement of other organs (e.g., lungs), the problem is more complex in the case of isolated abdominal SA. The recent use of contrast-enhanced ultrasound and endoscopic ultrasound elastography has provided additional information about the enhancement patterns and tissue rigidity in abdominal SA. For more information click here.
 Satoyoshi syndrome is a multisystemic rare disease of unknown etiology, although an autoimmune basis is presumed. Its main symptoms are: painful muscle spasms, diarrhea, alopecia and skeletal abnormalities. Clinical course without treatment may result in serious disability or death. A review of treatment and its response is still pending. Sixty-four cases of Satoyoshi syndrome were published between 1967 and 2018. 47 cases described the treatment administered. Drugs used can be divided into two main groups of treatment: muscle relaxants/anticonvulsants, and corticosteroids/
Satoyoshi syndrome is a multisystemic rare disease of unknown etiology, although an autoimmune basis is presumed. Its main symptoms are: painful muscle spasms, diarrhea, alopecia and skeletal abnormalities. Clinical course without treatment may result in serious disability or death. A review of treatment and its response is still pending. Sixty-four cases of Satoyoshi syndrome were published between 1967 and 2018. 47 cases described the treatment administered. Drugs used can be divided into two main groups of treatment: muscle relaxants/anticonvulsants, and corticosteroids/
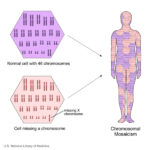 Turner syndrome is a rare condition in women that is associated with either complete or partial loss of one X chromosome, often in mosaic karyotypes. Turner syndrome is associated with short stature, delayed puberty, ovarian dysgenesis, hypergonadotropic hypogonadism, infertility, congenital malformations of the heart, endocrine disorders such as type 1 and type 2 diabetes mellitus, osteoporosis and autoimmune disorders. Morbidity and mortality are increased in women with Turner syndrome compared with the general population and the involvement of multiple organs through all stages of life necessitates a multidisciplinary approach to care. Despite an often conspicuous phenotype, the diagnostic delay can be substantial and the average age at diagnosis is around 15 years of age. However, numerous important clinical advances have been achieved, covering all specialty fields involved in the care of girls and women with Turner syndrome. Here, we present an updated Review of Turner syndrome, covering advances in genetic and genomic mechanisms of disease, associated disorders and multidisciplinary approaches to patient management, including growth hormone therapy and hormone replacement therapy. For more information click here.
Turner syndrome is a rare condition in women that is associated with either complete or partial loss of one X chromosome, often in mosaic karyotypes. Turner syndrome is associated with short stature, delayed puberty, ovarian dysgenesis, hypergonadotropic hypogonadism, infertility, congenital malformations of the heart, endocrine disorders such as type 1 and type 2 diabetes mellitus, osteoporosis and autoimmune disorders. Morbidity and mortality are increased in women with Turner syndrome compared with the general population and the involvement of multiple organs through all stages of life necessitates a multidisciplinary approach to care. Despite an often conspicuous phenotype, the diagnostic delay can be substantial and the average age at diagnosis is around 15 years of age. However, numerous important clinical advances have been achieved, covering all specialty fields involved in the care of girls and women with Turner syndrome. Here, we present an updated Review of Turner syndrome, covering advances in genetic and genomic mechanisms of disease, associated disorders and multidisciplinary approaches to patient management, including growth hormone therapy and hormone replacement therapy. For more information click here.
 Diamond-Blackfan anemia is a rare congenital red blood cell aplasia characterized by failed erythropoiesis, congenital abnormalities in up to 50% of patients, growth retardation in up to 30% of patients, and a predisposition to malignancy. Diamond-Blackfan anemia is both clinically and genetically a heterogenous condition ranging from subtle asymptomatic erythroid abnormalities to non-immune hydrops fetalis. Current treatment options include corticosteroid therapy, chronic red blood cell transfusions, and hematopoietic stem cell transplantation with gene therapy receiving recent attention. We report the first documented case of Diamond-Blackfan anemia in a Caucasian girl secondary to a sporadic heterozygous whole gene deletion in RPL35A in South Africa. Limited resources, non-availability of tests, unfamiliarity that comes with rare diseases, an expanded differential diagnosis, and an associated neutropenia led to a delay in the diagnosis of Diamond-Blackfan anemia. This case reminds clinicians of Diamond-Blackfan anemia as a cause of aplastic anemia and highlights the difficulty and obstacles in diagnosing Diamond-Blackfan anemia in resource-limited countries. Diamond-Blackfan anemia is a rare disease that carries significant morbidity and mortality if not diagnosed early and managed appropriately. Limited health resources, patient registries, and specialists as seen in developing countries result in a paucity of knowledge about Diamond-Blackfan anemia in Africa. This case reminds clinicians about Diamond-Blackfan anemia as a cause for anemia in infants, the limitations in making the diagnosis in under-resourced health care systems, and the need for standardized treatment protocols applicable to resource-limited countries. For more information click here.
Diamond-Blackfan anemia is a rare congenital red blood cell aplasia characterized by failed erythropoiesis, congenital abnormalities in up to 50% of patients, growth retardation in up to 30% of patients, and a predisposition to malignancy. Diamond-Blackfan anemia is both clinically and genetically a heterogenous condition ranging from subtle asymptomatic erythroid abnormalities to non-immune hydrops fetalis. Current treatment options include corticosteroid therapy, chronic red blood cell transfusions, and hematopoietic stem cell transplantation with gene therapy receiving recent attention. We report the first documented case of Diamond-Blackfan anemia in a Caucasian girl secondary to a sporadic heterozygous whole gene deletion in RPL35A in South Africa. Limited resources, non-availability of tests, unfamiliarity that comes with rare diseases, an expanded differential diagnosis, and an associated neutropenia led to a delay in the diagnosis of Diamond-Blackfan anemia. This case reminds clinicians of Diamond-Blackfan anemia as a cause of aplastic anemia and highlights the difficulty and obstacles in diagnosing Diamond-Blackfan anemia in resource-limited countries. Diamond-Blackfan anemia is a rare disease that carries significant morbidity and mortality if not diagnosed early and managed appropriately. Limited health resources, patient registries, and specialists as seen in developing countries result in a paucity of knowledge about Diamond-Blackfan anemia in Africa. This case reminds clinicians about Diamond-Blackfan anemia as a cause for anemia in infants, the limitations in making the diagnosis in under-resourced health care systems, and the need for standardized treatment protocols applicable to resource-limited countries. For more information click here.
 Hemophilia A (HA) is an X-linked hereditary bleeding disorder caused by deficiency of coagulation factor (F) VIII activity. One of the greatest complications in the treatment of HA is the development of neutralizing alloantibodies, known as FVIII inhibitors. HA patients who develop FVIII inhibitors have limited treatment options available to them and experience greater disease- and treatment-related burdens than HA patients without FVIII inhibitors. Emicizumab, a recently approved bispecific monoclonal antibody, mimics the function of FVIIIa by bridging FIXa and FX to restore effective hemostasis. Although emicizumab and FVIII show some functional similarities, several key differences influence the results of standard laboratory assays when conducted in the presence of emicizumab, and can result in a misleading interpretation of coagulation assays in emicizumab-treated patients. Here, we discuss current laboratory monitoring methods, including activated partial thromboplastin time, FVIII one-stage clotting assays, FVIII chromogenic assays, and global coagulations assays; address why these conventional methods may be inappropriate for monitoring of HA patients receiving emicizumab; and suggest alternative methods applicable to monitoring HA treatment in an evolving treatment landscape. For more information click here.
Hemophilia A (HA) is an X-linked hereditary bleeding disorder caused by deficiency of coagulation factor (F) VIII activity. One of the greatest complications in the treatment of HA is the development of neutralizing alloantibodies, known as FVIII inhibitors. HA patients who develop FVIII inhibitors have limited treatment options available to them and experience greater disease- and treatment-related burdens than HA patients without FVIII inhibitors. Emicizumab, a recently approved bispecific monoclonal antibody, mimics the function of FVIIIa by bridging FIXa and FX to restore effective hemostasis. Although emicizumab and FVIII show some functional similarities, several key differences influence the results of standard laboratory assays when conducted in the presence of emicizumab, and can result in a misleading interpretation of coagulation assays in emicizumab-treated patients. Here, we discuss current laboratory monitoring methods, including activated partial thromboplastin time, FVIII one-stage clotting assays, FVIII chromogenic assays, and global coagulations assays; address why these conventional methods may be inappropriate for monitoring of HA patients receiving emicizumab; and suggest alternative methods applicable to monitoring HA treatment in an evolving treatment landscape. For more information click here.
 Rare ocular pathology has an important impact on the quality of life of patients because often the damage is bilateral and, although asymmetric, causes a significant decrease in visual acuity. Because it may be asymptomatic until a relatively late stage, diagnosis is frequently delayed. A general understanding of the disease pathophysiology, diagnosis, and treatment may assist primary care physicians in referring high-risk patients for comprehensive ophthalmological examination and for a more active involvement in their care. Moreover, a significant percentage of these orphan diseases do not have treatment approved by the FDA. The examination and monitoring of patients with rare ophthalmological disorders represents a key component of an ongoing project at the University Emergency Hospital, Bucharest, Romania – Ophthalmology Clinic. Rare disease registries are leading tools for the development of clinical research for rare diseases, improvement of patient access to new diagnostic methods, follow-up and new emerging therapies. As of this moment, the European list of rare diseases includes 53 ophthalmological diseases, which are classified as rare diseases and another 103 systemic diseases with ophthalmological involvement, out of a total of 7000 rare diseases. For more information click here.
Rare ocular pathology has an important impact on the quality of life of patients because often the damage is bilateral and, although asymmetric, causes a significant decrease in visual acuity. Because it may be asymptomatic until a relatively late stage, diagnosis is frequently delayed. A general understanding of the disease pathophysiology, diagnosis, and treatment may assist primary care physicians in referring high-risk patients for comprehensive ophthalmological examination and for a more active involvement in their care. Moreover, a significant percentage of these orphan diseases do not have treatment approved by the FDA. The examination and monitoring of patients with rare ophthalmological disorders represents a key component of an ongoing project at the University Emergency Hospital, Bucharest, Romania – Ophthalmology Clinic. Rare disease registries are leading tools for the development of clinical research for rare diseases, improvement of patient access to new diagnostic methods, follow-up and new emerging therapies. As of this moment, the European list of rare diseases includes 53 ophthalmological diseases, which are classified as rare diseases and another 103 systemic diseases with ophthalmological involvement, out of a total of 7000 rare diseases. For more information click here.
 For the tenth consecutive year, the Institute for Rare Diseases and Orphan Drugs organises a national conference. This year the conference will be held under the patronage of the Health Commission at the 44th National Assembly of the Republic of Bulgaria.
For the tenth consecutive year, the Institute for Rare Diseases and Orphan Drugs organises a national conference. This year the conference will be held under the patronage of the Health Commission at the 44th National Assembly of the Republic of Bulgaria.
“Within its remit, the Health Commission has always been an open partner to support the efforts of institutions and non-governmental organizations to improve the development of prevention, diagnosis, treatment, rehabilitation and social care for people with rare diseases,” the letter said Dr. Daniela Daritkova.
The main topic of the event will be the novelties and current trends in the diagnosis, treatment and follow-up of rare diseases, the development of European reference networks and access to innovation in the field of rare diseases.
The 15th anniversary of the Information Center for Rare Diseases and Orphan Drugs (ICRDOD) will also be celebrated during the event. The Information Center started its activity in 2004 as the first information center of its kind in Eastern Europe dedicated to patients, organizations and medical specialists with an interest in rare diseases and orphan drugs. ICRDOD has been developing over the past 15 years with the support and assistance of 26 volunteer consultants – leading medical specialists from the university clinics in Bulgaria. For more information click here.
 Achieving normal growth is one of the most challenging problems in the management of children with chronic kidney disease (CKD). Treatment with recombinant human growth hormone (GH) promotes longitudinal growth and likely enables children with CKD and short stature to reach normal adult height. Here, members of the European Society for Paediatric Nephrology (ESPN) CKD-Mineral and Bone Disorder (MBD), Dialysis and Transplantation working groups present clinical practice recommendations for the use of GH in children with CKD on dialysis and after renal transplantation. These recommendations have been developed with input from an external advisory group of paediatric endocrinologists, paediatric nephrologists and patient representatives. We recommend that children with stage 3-5 CKD or on dialysis should be candidates for GH therapy if they have persistent growth failure, defined as a height below the third percentile for age and sex and a height velocity below the twenty-fifth percentile, once other potentially treatable risk factors for growth failure have been adequately addressed and provided the child has growth potential. In children who have received a kidney transplant and fulfil the above growth criteria, we recommend initiation of GH therapy 1 year after transplantation if spontaneous catch-up growth does not occur and steroid-free immunosuppression is not a feasible option. GH should be given at dosages of 0.045-0.05 mg/kg per day by daily subcutaneous injections until the patient has reached their final height or until renal transplantation. In addition to providing treatment recommendations, a cost-effectiveness analysis is provided that might help guide decision-making. For more information click here.
Achieving normal growth is one of the most challenging problems in the management of children with chronic kidney disease (CKD). Treatment with recombinant human growth hormone (GH) promotes longitudinal growth and likely enables children with CKD and short stature to reach normal adult height. Here, members of the European Society for Paediatric Nephrology (ESPN) CKD-Mineral and Bone Disorder (MBD), Dialysis and Transplantation working groups present clinical practice recommendations for the use of GH in children with CKD on dialysis and after renal transplantation. These recommendations have been developed with input from an external advisory group of paediatric endocrinologists, paediatric nephrologists and patient representatives. We recommend that children with stage 3-5 CKD or on dialysis should be candidates for GH therapy if they have persistent growth failure, defined as a height below the third percentile for age and sex and a height velocity below the twenty-fifth percentile, once other potentially treatable risk factors for growth failure have been adequately addressed and provided the child has growth potential. In children who have received a kidney transplant and fulfil the above growth criteria, we recommend initiation of GH therapy 1 year after transplantation if spontaneous catch-up growth does not occur and steroid-free immunosuppression is not a feasible option. GH should be given at dosages of 0.045-0.05 mg/kg per day by daily subcutaneous injections until the patient has reached their final height or until renal transplantation. In addition to providing treatment recommendations, a cost-effectiveness analysis is provided that might help guide decision-making. For more information click here.
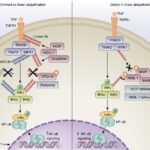 Monogenic autoinflammatory disorders (AIDs) are rare diseases caused by variants in genes regulating the innate immune system. The identification of the first four genes responsible for the prototype group of hereditary recurrent fevers prompted the development of genetic diagnosis, followed by external quality assessment and guidelines for the interpretation of sequence variants in these diseases. Recent changes in the diagnosis of genetic diseases, namely the implementation of next-generation sequencing (NGS), lead to discovery of the new genes associated with at least 40 novel AIDs, which revolutionized patient care and prognosis. However, these rapid advances resulted in nonstandardized molecular strategies that can influence genetic diagnosis and reporting of results. In order to assess factors, which may have an impact on performance and quality of results in the NGS era, we carried out an online survey among member laboratories of the European Molecular Genetics Quality Network, which highlighted different strategies being used and identified pitfalls that deserve discussion and improvement. For more information click here.
Monogenic autoinflammatory disorders (AIDs) are rare diseases caused by variants in genes regulating the innate immune system. The identification of the first four genes responsible for the prototype group of hereditary recurrent fevers prompted the development of genetic diagnosis, followed by external quality assessment and guidelines for the interpretation of sequence variants in these diseases. Recent changes in the diagnosis of genetic diseases, namely the implementation of next-generation sequencing (NGS), lead to discovery of the new genes associated with at least 40 novel AIDs, which revolutionized patient care and prognosis. However, these rapid advances resulted in nonstandardized molecular strategies that can influence genetic diagnosis and reporting of results. In order to assess factors, which may have an impact on performance and quality of results in the NGS era, we carried out an online survey among member laboratories of the European Molecular Genetics Quality Network, which highlighted different strategies being used and identified pitfalls that deserve discussion and improvement. For more information click here.
 Among angio-oedema patients, hereditary angio-oedema (HAE) should not be overlooked. Besides skin swellings, these patients might have very painful abdominal attacks and potentially life-threatening angio-oedema of the upper airway. They will not respond to traditional anti-allergic therapy with antihistamines, corticosteroids, and adrenaline, and instead need specific drugs targeting the kallikrein-kinin pathway. Classically, patients with HAE have a quantitative or qualitative deficiency of the C1 inhibitor (C1INH) due to different mutations in SERPING1, although a new subtype with normal C1INH has been recognised more recently. This latter variant is diagnosed based on clinical features, family history, or molecular genetic testing for mutations in F12, ANGPT1,or PLG.The diagnosis of HAE is often delayed due to a general unfamiliarity with this orphan disease. However, undiagnosed patients are at an increased risk of unnecessary surgical interventions or life-threatening laryngeal swellings. Within the last decade, new and effective therapies have been developed and launched for acute and prophylactic therapy. Even more drugs are under evaluation in clinical trials. It is therefore of utmost importance that patients with HAE are diagnosed as soon as possible and offered relevant therapy with orphan drugs to reduce morbidity, prevent mortality, and improve quality of life. For more information click here.
Among angio-oedema patients, hereditary angio-oedema (HAE) should not be overlooked. Besides skin swellings, these patients might have very painful abdominal attacks and potentially life-threatening angio-oedema of the upper airway. They will not respond to traditional anti-allergic therapy with antihistamines, corticosteroids, and adrenaline, and instead need specific drugs targeting the kallikrein-kinin pathway. Classically, patients with HAE have a quantitative or qualitative deficiency of the C1 inhibitor (C1INH) due to different mutations in SERPING1, although a new subtype with normal C1INH has been recognised more recently. This latter variant is diagnosed based on clinical features, family history, or molecular genetic testing for mutations in F12, ANGPT1,or PLG.The diagnosis of HAE is often delayed due to a general unfamiliarity with this orphan disease. However, undiagnosed patients are at an increased risk of unnecessary surgical interventions or life-threatening laryngeal swellings. Within the last decade, new and effective therapies have been developed and launched for acute and prophylactic therapy. Even more drugs are under evaluation in clinical trials. It is therefore of utmost importance that patients with HAE are diagnosed as soon as possible and offered relevant therapy with orphan drugs to reduce morbidity, prevent mortality, and improve quality of life. For more information click here.