
 The aim is to describe the specifics of health technology assessment (HTA) of targeted therapies and discuss important issues related to current research methods and application of HTA process in Bulgaria. Analytic study based on literature review and content analysis of HTA guidelines and Bulgarian HTA legislation wеre performed. Specifics of HTA of targeted therapies were analyzed through determining the characteristics of the object and the subject of analysis. Characteristics and types of targeted therapies were defined. Specific features and challenges of HTA of targeted therapies were discussed. The issue of companion diagnostic tests associated with targeted therapies was highlighted. There are important methodological issues of HTA of targeted therapies determined by the object of analysis that should be taken into consideration both in the process of assessment and appraisal of targeted therapies in Bulgaria. For more information click here.
The aim is to describe the specifics of health technology assessment (HTA) of targeted therapies and discuss important issues related to current research methods and application of HTA process in Bulgaria. Analytic study based on literature review and content analysis of HTA guidelines and Bulgarian HTA legislation wеre performed. Specifics of HTA of targeted therapies were analyzed through determining the characteristics of the object and the subject of analysis. Characteristics and types of targeted therapies were defined. Specific features and challenges of HTA of targeted therapies were discussed. The issue of companion diagnostic tests associated with targeted therapies was highlighted. There are important methodological issues of HTA of targeted therapies determined by the object of analysis that should be taken into consideration both in the process of assessment and appraisal of targeted therapies in Bulgaria. For more information click here.
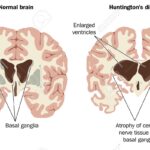 Huntington’s disease (HD) is a rare, hereditary, neurodegenerative and dominantly transmitted disorder affecting about 10 out of 100,000 people in Western Countries. The genetic cause is a CAG repeat expansion in the huntingtin gene (HTT), which is unstable and may further increase its length in subsequent generations, so called anticipation. Mutation repeat length coupled with other gene modifiers and environmental factors contribute to the age at onset in the offspring. Considering the unpredictability of age at onset and of clinical prognosis in HD, the accurate interpretation, a proper psychological support and a scientifically sound and compassionate communication of the genetic test result are crucial in the context of Good Clinical Practice and when considering further potential disease-modifying therapies. We discuss various genetic test scenarios that require a particularly careful attention in psychological and genetic counseling and expect that the counseling procedures will require a constant update. For more information click here.
Huntington’s disease (HD) is a rare, hereditary, neurodegenerative and dominantly transmitted disorder affecting about 10 out of 100,000 people in Western Countries. The genetic cause is a CAG repeat expansion in the huntingtin gene (HTT), which is unstable and may further increase its length in subsequent generations, so called anticipation. Mutation repeat length coupled with other gene modifiers and environmental factors contribute to the age at onset in the offspring. Considering the unpredictability of age at onset and of clinical prognosis in HD, the accurate interpretation, a proper psychological support and a scientifically sound and compassionate communication of the genetic test result are crucial in the context of Good Clinical Practice and when considering further potential disease-modifying therapies. We discuss various genetic test scenarios that require a particularly careful attention in psychological and genetic counseling and expect that the counseling procedures will require a constant update. For more information click here.
Neural tube defects (NTD’s) represent a group of severe congenital abnormalities that are associated with high mortality, adverse outcomes and long-term disabilities, as well as significant emotional, psychological and economic consequences. Every year, approximately 5 000 cases of NTD’s occur in the EU. Most of them are diagnosed prenatally with subsequent termination of pregnancy. Preconceptual intake of folic acid has been found to significantly reduce the risk for NTD’s. Nevertheless, to date, there are no folic acid programs available in European countries for women in reproductive age or, in particular, for those who are planning a pregnancy. The publication aims to present and analyze new trends in the use of folic acid as a primary prevention of rare genetic diseases. For more information click here.
Synonym(s):Epilepsy-dementia-amelogenesis imperfecta syndrome
Kohlschutter-Tonz syndrome
ICD-10: G40.8
ORPHANET number: ORPHA1946
For more information about this disease, please visit the Orphanet website.
Visitors from North America may also contact NIH Office of Rare Diseases Research website.
—————————————————————————
Last modification: 12:40 21.05.2024
—————————————————————————
 Over the last decade, several large registries of patients with IPF have been established. These are collecting a wealth of longitudinal data on thousands of patients with this rare disease. The data collected in these registries will be complementary to those collected in clinical trials, as the patient populations studied in registries have a broader spectrum of disease severity and comorbidities and can be followed for a longer period of time. Maintaining the quality and completeness of registry databases presents administrative and resourcing challenges, but is important to ensure the robustness of the analyses. Data from patient registries have already helped improve understanding of the clinical characteristics of patients with IPF, the impact that the disease has on their quality of life and survival, and current practices in diagnosis and management. In the future, analyses of biospecimens linked to detailed patient profiles will provide the opportunity to identify biomarkers linked to disease progression, facilitating the development of precision medicine approaches for prognosis and therapy in patients with IPF. For more information click here.
Over the last decade, several large registries of patients with IPF have been established. These are collecting a wealth of longitudinal data on thousands of patients with this rare disease. The data collected in these registries will be complementary to those collected in clinical trials, as the patient populations studied in registries have a broader spectrum of disease severity and comorbidities and can be followed for a longer period of time. Maintaining the quality and completeness of registry databases presents administrative and resourcing challenges, but is important to ensure the robustness of the analyses. Data from patient registries have already helped improve understanding of the clinical characteristics of patients with IPF, the impact that the disease has on their quality of life and survival, and current practices in diagnosis and management. In the future, analyses of biospecimens linked to detailed patient profiles will provide the opportunity to identify biomarkers linked to disease progression, facilitating the development of precision medicine approaches for prognosis and therapy in patients with IPF. For more information click here.
A retrospective analysis of direct costs for intravenous bisphosphonate (BF) therapy for the period 2012-2016 is done. Data are derived from the registers of the pharmacies at the hospitals in Plovdiv District where BF therapy is applied. In addition, the annual costs for zoledronic acid available from the NHIF register are also analyzed. All costs are quantified on the basis of national rates set by the legislator. The direct cost of drug therapy for the country falls 32 times from 2018 to 2012 – from BGN 8 983 283.42 to BGN 276 722.51. This trend is also observed in the data from hospital pharmacies where the drop is 99 times from 2012 to 2016 – BGN 2,334,138.90 to BGN 23,542.65, but not only the decrease in the cost of therapy, but also 4 times smaller number of packages paid – from 4004 pcs. for 2012 to 1402 pcs. for the year 2016. An important consequence of the illness is that individuals can not perform their usual daily activities and so, besides spending the NHIF’s budget, they suffer production losses. For more information click here.
 Diagnosis of inherited bleeding disorders (IBDs) remains challenging, especially in the case of inherited platelet disorders, due to the heterogeneity of the clinical and laboratory phenotype, the limited specificity of platelet function tests, and the large number of potential culprit genes. Nowadays, high-throughput sequencing (HTS) allows the simultaneous and rapid investigation of multiple genes at a manageable cost. This HTS technology that includes targeted sequencing of prespecified genes, whole-exome sequencing, or whole-genome sequencing, is revolutionizing the genetic diagnosis of human diseases. Through its extensive implementation in research and clinical practice, HTS is rapidly improving the molecular characterization of IBDs. However, despite the availability of this powerful approach, many patients still do not receive a diagnosis. As IBDs are complex and rare diseases, development of more advanced laboratory assays, improvements in bioinformatic pipelines, and the formation of multidisciplinary teams are encouraged to advance our understanding of IBDs. For more information click here.
Diagnosis of inherited bleeding disorders (IBDs) remains challenging, especially in the case of inherited platelet disorders, due to the heterogeneity of the clinical and laboratory phenotype, the limited specificity of platelet function tests, and the large number of potential culprit genes. Nowadays, high-throughput sequencing (HTS) allows the simultaneous and rapid investigation of multiple genes at a manageable cost. This HTS technology that includes targeted sequencing of prespecified genes, whole-exome sequencing, or whole-genome sequencing, is revolutionizing the genetic diagnosis of human diseases. Through its extensive implementation in research and clinical practice, HTS is rapidly improving the molecular characterization of IBDs. However, despite the availability of this powerful approach, many patients still do not receive a diagnosis. As IBDs are complex and rare diseases, development of more advanced laboratory assays, improvements in bioinformatic pipelines, and the formation of multidisciplinary teams are encouraged to advance our understanding of IBDs. For more information click here.
 TK2 gene encodes for mitochondrial thymidine kinase, which phosphorylates the pyrimidine nucleosides thymidine and deoxycytidine. Recessive mutations in the TK2 gene are responsible for the ‘myopathic form’ of the mitochondrial depletion/multiple deletions syndrome, with a wide spectrum of severity. We describe 18 patients with mitochondrial myopathy due to mutations in the TK2 gene with absence of clinical symptoms until the age of 12. The mean age of onset was 31 years. The first symptom was muscle limb weakness in 10/18, eyelid ptosis in 6/18, and respiratory insufficiency in 2/18. All patients developed variable muscle weakness during the evolution of the disease. Half of patients presented difficulty in swallowing. All patients showed evidence of respiratory muscle weakness, with need for non-invasive Mechanical Ventilation in 12/18. The late-onset is the less frequent form of presentation of the TK2 deficiency and its natural history is not well known. Patients with late onset TK2 deficiency have a consistent and recognizable clinical phenotype and a poor prognosis, due to the high risk of early and progressive respiratory insufficiency. For more information click here.
TK2 gene encodes for mitochondrial thymidine kinase, which phosphorylates the pyrimidine nucleosides thymidine and deoxycytidine. Recessive mutations in the TK2 gene are responsible for the ‘myopathic form’ of the mitochondrial depletion/multiple deletions syndrome, with a wide spectrum of severity. We describe 18 patients with mitochondrial myopathy due to mutations in the TK2 gene with absence of clinical symptoms until the age of 12. The mean age of onset was 31 years. The first symptom was muscle limb weakness in 10/18, eyelid ptosis in 6/18, and respiratory insufficiency in 2/18. All patients developed variable muscle weakness during the evolution of the disease. Half of patients presented difficulty in swallowing. All patients showed evidence of respiratory muscle weakness, with need for non-invasive Mechanical Ventilation in 12/18. The late-onset is the less frequent form of presentation of the TK2 deficiency and its natural history is not well known. Patients with late onset TK2 deficiency have a consistent and recognizable clinical phenotype and a poor prognosis, due to the high risk of early and progressive respiratory insufficiency. For more information click here.
Gender differences in clinical presentation and vascular pattern in patients with Takayasu arteritis
 To compare clinical characteristics and pattern of vascular involvement at disease onset according to gender specificity in patients with Takayasu arteritis (TA). Data from 117 TA patients (11 male, 106 female), diagnosed according to the American College of Rheumatology criteria, from our centre were retrospectively collected. Differences between men and women regarding demographic features, diagnostic delay, signs and symptoms attributed to TA, and arteries involved at diagnosis were compared. Data were obtained from three published articles describing gender differences in TA. A global analysis of these three cohorts plus ours (a total of 578 patients; 108 men, 470 women) was performed. In our TA cohort, age at disease onset and age at diagnosis were not significantly different between genders. Diagnostic delay was higher in men. Male patients showed higher involvement of iliac arteries, females suffered more frequently from upper limb claudication. In TA patients, gender has a strong influence on pattern of vascular involvement and consequently on clinical presentation. Specifically, women have a higher involvement of the supradiaphragmatic vessels, whereas in men the abdominal vessels are more frequently affected. For more information click here.
To compare clinical characteristics and pattern of vascular involvement at disease onset according to gender specificity in patients with Takayasu arteritis (TA). Data from 117 TA patients (11 male, 106 female), diagnosed according to the American College of Rheumatology criteria, from our centre were retrospectively collected. Differences between men and women regarding demographic features, diagnostic delay, signs and symptoms attributed to TA, and arteries involved at diagnosis were compared. Data were obtained from three published articles describing gender differences in TA. A global analysis of these three cohorts plus ours (a total of 578 patients; 108 men, 470 women) was performed. In our TA cohort, age at disease onset and age at diagnosis were not significantly different between genders. Diagnostic delay was higher in men. Male patients showed higher involvement of iliac arteries, females suffered more frequently from upper limb claudication. In TA patients, gender has a strong influence on pattern of vascular involvement and consequently on clinical presentation. Specifically, women have a higher involvement of the supradiaphragmatic vessels, whereas in men the abdominal vessels are more frequently affected. For more information click here.
 Rett syndrome (RTT) is an early-onset neurodevelopmental disorder that is caused by mutations in the MECP2 gene; however, defects in other genes (CDKL5 and FOXG1) can lead to presentations that resemble classic RTT, although they are not completely identical. Here, we attempted to identify other monogenic disorders that share features of RTT. A total of 437 patients with a clinical diagnosis of RTT-like were studied. A total of 40 patients with clinical features of RTT had variants which affect gene function in six genes associated with other monogenic disorders Genetic studies using next generation sequencing (NGS) allowed us to study a larger number of genes associated with RTT-like simultaneously, providing a genetic diagnosis for a wider group of patients. These new findings provide the clinician with more information and clues that could help in the prevention of future symptoms or in pharmacologic therapy. For more information click here.
Rett syndrome (RTT) is an early-onset neurodevelopmental disorder that is caused by mutations in the MECP2 gene; however, defects in other genes (CDKL5 and FOXG1) can lead to presentations that resemble classic RTT, although they are not completely identical. Here, we attempted to identify other monogenic disorders that share features of RTT. A total of 437 patients with a clinical diagnosis of RTT-like were studied. A total of 40 patients with clinical features of RTT had variants which affect gene function in six genes associated with other monogenic disorders Genetic studies using next generation sequencing (NGS) allowed us to study a larger number of genes associated with RTT-like simultaneously, providing a genetic diagnosis for a wider group of patients. These new findings provide the clinician with more information and clues that could help in the prevention of future symptoms or in pharmacologic therapy. For more information click here.