
СТАРТИРА ИНФОРМАЦИОННАТА КАМПАНИЯ „6000 ПРИЧИНИ ЗА ДЕЙСТВИЕ“ В ПОДКРЕПА НА ПАЦИЕНТИТЕ С РЕДКИ БОЛЕСТИ В БЪЛГАРИЯ
Навременна диагностика, достъп до съвременно лечение, подкрепа и интеграция за пациентите, както и въвеждане на Национален план за редките болести са сред целите на инициативата

В навечерието на Световния ден на редките болести – 29 февруари 2024 г., стартира информационна кампания „6000 причини за действие“. Инициативата има за цел да повиши информираността на обществото по темата за редките болести, както и да постави ключови въпроси, свързани с подобряване на възможностите за навременна диагностика, достъпа до съвременно лечение на пациентите и приемане на Национален план за редките болести в България, който да обезпечи нуждите на обществото и медицинската общност в тази сфера.
Кампанията се организира от Асоциацията на научноизследователските фармацевтични производители в България (ARPharM) и се провежда под патронажа на Комисията по здравеопазването към 49-то Народно събрание. Партньори на кампанията са Министерството на здравеопазването, Националната здравноосигурителна каса, Българският лекарски съюз, Националната генетична лаборатория, Българската педиатрична асоциация, Българската асоциация по неонатология, Българското дружество по алергология, Българското дружество по нефрология, Българското дружество по невромускулни заболявания, Българското дружество по детска гастроентерология, хепатология и хранене, Българското дружество по детска офталмология, невроофталмология и офталмогенетика, Българското медицинско дружество по детска хематология и онкология, Варненското педиатрично ендокринологично дружество, Асоциацията на студентите-медици в България – София, Институтът по редки болести, Националният алианс на хората с редки болести, Българската асоциация за закрила на пациентите, Сдружението за развитие на българското здравеопазване, Асоциацията „Наследствен ангиоедем“, Фондацията „Знанието дарява живот“ и Националното сдружение на общините в Република България.
В момента в света са открити над 6000 редки болести, като 72% от тях са генетични и близо 70% се проявяват още в детска възраст. За рядко се счита всяко заболяване, което засяга между един и пет на 10 000 души население или до двама на 2000 души. Всички известни до момента редки заболявания засягат средно между 3,5-5,9% от населението в световен мащаб или повече от 300 млн.[1] души.
В България са регистрирани едва 200 редки заболявания, като поради липса на регистри е трудно да се каже какъв е реалният брой на пациентите. Смята се, че редките болести засягат повече от 350 000 българи. В същото време, заради липсата на скринингови програми и късното диагностициране, се предполага, че в България живеят множество пациенти, страдащи от рядко заболяване, които все още не знаят това. Към момента масовият неонатален скрининг при новородени включва само 3 заболявания: фенилкетонурия, надбъбречнокорова хиперплазия и вроден хипотиреоидизъм. Така страната ни се нарежда на едно от последните места по обхват на неонаталния скрининг не само в Европейския съюз, но и в Европа. Разширяването на скрининговия панел при новородени и деца е съществена стъпка към повишаване на ранното диагностициране на редките заболявания.
Друг съществен проблем представляват диагностичните изследвания за редки заболявания. Експертните институции и специализираните центрове за диагностика и лечение на редки заболявания са малко на брой, не са добре познати или са трудно достъпни. За доказването на определена рядка диагноза са необходими множество изследвания, сред които и скъпоструващи генетични тестове. Същевременно достъпът до тях е силно затруднен и пациентите и техните семейства са принудени да ги заплащат със собствени средства.
Не на последно място стои въпросът с достъпа до лечение. От регистрираните в Европа за периода 2018-2021 г. 61 лекарства-сираци в България са налични едва 11, което е много под средното за ЕС (24 продукта) и далеч след Германия (55 продукта) и Италия (50 продукта). В същото време българските пациенти имат достъп до лекарства-сираци 858 дни след разрешението за употреба. Това е почти най-дългият период на забавяне на достъпа до лечение. Средното време за ЕС е 548 дни, а държавите с най-бърз достъп са Германия (45 дни) и Австрия (99 дни)[2]. Фондът за лечение на деца предоставя частично решение за педиатричните пациенти с редки заболявания, но при навършване на 18 години съществува риск достъпът до лечение да бъде прекъснат, в случай че съответният лекарствен продукт не фигурира в Позитивния лекарствен списък.
Изброените проблеми, както и необходимостта от политики за психо-социална адаптация и подкрепа на пациентите и техните близки, актуализиране и допълнение на обучителните програми за специалистите и др., налагат спешното актуализиране на Национален план за редки болести, което в последното си издание е с обхват 2009-2013 г. Необходими са систематични и целенасочени политики, за да бъдат преодолени описаните дефицити в здравната система, с което да се подобрят грижите за пациентите с редки заболявания в България.
Информационната кампания в подкрепа на пациентите с редки болести „6000 причини за действие“ ще бъде дигитална и ще се реализира в социалните мрежи Facebook (www.facebook.com/WeWontRest) и LinkedIn (www.linkedin.com/company/niamadasprem/). На страниците на кампанията ще бъдат публикувани видеа с пациентски истории, които ще представят трудния път от диагнозата до лечението. На Световния ден на редките заболявания (29 февруари 2024 г.) от 19:00 до 20:00 ч. ще бъдат осветени емблематични сгради в София и страната, като инициативата се осъществява с подкрепата на Националното сдружение на общините в Република България. В столицата в цветовете, символизиращи глобалната инициатива за отбелязване на Световния ден, ще бъде осветен Националният дворец на културата. Чрез специално видео, излъчвано в столичното метро, пациенти с редки заболявания ще разкажат за предизвикателствата, с които ежедневно се сблъскват, още от самото поставяне на диагнозата, до получаване на необходимата терапия и възможността за последваща грижа и подкрепа за тях и техните семейства.
Всички информационни материали, в това число инфографики, видео клипове с пациентски истории и данни за редките заболявания, можете да откриете на: www.facebook.com/WeWontRest и www.linkedin.com/company/niamadasprem/
Основното видео на кампанията можете да видите на:
[1] https://www.rarediseaseday.org/
[2] EFPIA Patients W.A.I.T. Indicator 2022 Survey, Published April 2023
 Синдромът на Фанкони-Бикел (ФБС) е рядко автозомно рецесивно заболяване, характеризиращо се с нарушена утилизация на глюкоза и галактоза, както и проксимална бъбречна тубулна дисфункция. В това проучване са анализирани ретроспективно клинични, биохимични, генетични данни, данни за лечение и проследяване на 11 педиатрични пациенти с ФБС.
Синдромът на Фанкони-Бикел (ФБС) е рядко автозомно рецесивно заболяване, характеризиращо се с нарушена утилизация на глюкоза и галактоза, както и проксимална бъбречна тубулна дисфункция. В това проучване са анализирани ретроспективно клинични, биохимични, генетични данни, данни за лечение и проследяване на 11 педиатрични пациенти с ФБС. Приблизително 400 милиона души по света страдат от рядко заболяване. Въпреки че напредъкът в секвенирането на целия екзом и целия геном значително улесняват диагностицирането на редки заболявания, общите диагностични нива остават под 50%. Освен това, в случаите, когато се постига точна диагноза, процесът изисква средно 4,8 години. Намаляването на времето, необходимо за диагностика на заболяването, е сред най-критичните нужди на пациентите, засегнати от рядко заболяване.
Приблизително 400 милиона души по света страдат от рядко заболяване. Въпреки че напредъкът в секвенирането на целия екзом и целия геном значително улесняват диагностицирането на редки заболявания, общите диагностични нива остават под 50%. Освен това, в случаите, когато се постига точна диагноза, процесът изисква средно 4,8 години. Намаляването на времето, необходимо за диагностика на заболяването, е сред най-критичните нужди на пациентите, засегнати от рядко заболяване. Георги Искров, Вяра Ангелова, Боян Бочев, Васка Вълчинова, Теодора Генчева, Десислава Джулева, Юлиян Дичев, Таня Недкова, Мария Палкова, Анелия Тютюкова, Мария Христова, Елеонора Христова-Атанасова и Румен Стефанов, като част от Института по редки болести , катедра „Социална медицина и обществено здраве“ и Медицински факултет към Медицински университет – Пловдив проведоха всеобхватно проучване за гледните точки на специалисти по педиатрия, неонатология, медицинска генетика и биохимия за разширяване на обхвата на заболяванията, включени в масовия скрининг на новородени в България.
Георги Искров, Вяра Ангелова, Боян Бочев, Васка Вълчинова, Теодора Генчева, Десислава Джулева, Юлиян Дичев, Таня Недкова, Мария Палкова, Анелия Тютюкова, Мария Христова, Елеонора Христова-Атанасова и Румен Стефанов, като част от Института по редки болести , катедра „Социална медицина и обществено здраве“ и Медицински факултет към Медицински университет – Пловдив проведоха всеобхватно проучване за гледните точки на специалисти по педиатрия, неонатология, медицинска генетика и биохимия за разширяване на обхвата на заболяванията, включени в масовия скрининг на новородени в България. Наследственият ангиоедем с нормален C1-INH (HAE-nC1-INH) е рядко генетично заболяване с подобен фенотип на HAE-C1-INH, но с различен генетичен произход. В момента са известни шест подтипа, въз основа на причиняващите ги мутации.
Наследственият ангиоедем с нормален C1-INH (HAE-nC1-INH) е рядко генетично заболяване с подобен фенотип на HAE-C1-INH, но с различен генетичен произход. В момента са известни шест подтипа, въз основа на причиняващите ги мутации. За 15-та поредна година предстои да се проведе Национална конференция за редки болести и лекарства сираци, организирана от
За 15-та поредна година предстои да се проведе Национална конференция за редки болести и лекарства сираци, организирана от 
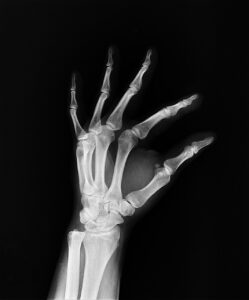 Това изследване има за цел да опише голяма група от италиански пациенти, засегнати от остеогенезис имперфекта, предоставяйки картина на клиничните костни и извънкостни характеристики и молекулярния фон, за да се подобрят познанията за заболяването и да се избере подходящо лечение в клиничната практика.
Това изследване има за цел да опише голяма група от италиански пациенти, засегнати от остеогенезис имперфекта, предоставяйки картина на клиничните костни и извънкостни характеристики и молекулярния фон, за да се подобрят познанията за заболяването и да се избере подходящо лечение в клиничната практика. В страните от Европейския съюз заболяване, което засяга по-малко от 5 души на 10 000, се счита за рядко. Поради недостига на опит и сложността на редките болести, пациентите могат да останат без диагноза години наред. Положени са усилия за включване на ORPHA – кодовете в здравно-информационните системи, което позволява кодирането на редки болести. Преди това тази номенклатура обхващаше само специфични редки заболявания, оставяйки тези с недиагностицирани състояния без кодове. Признаването на статут на редки болести е от решаващо значение за пациентите, подпомагайки възстановяването на разходите за грижи, осигурявайки социална и психологическа подкрепа и предоставяйки достъп до научни постижения, дори без точна диагноза.
В страните от Европейския съюз заболяване, което засяга по-малко от 5 души на 10 000, се счита за рядко. Поради недостига на опит и сложността на редките болести, пациентите могат да останат без диагноза години наред. Положени са усилия за включване на ORPHA – кодовете в здравно-информационните системи, което позволява кодирането на редки болести. Преди това тази номенклатура обхващаше само специфични редки заболявания, оставяйки тези с недиагностицирани състояния без кодове. Признаването на статут на редки болести е от решаващо значение за пациентите, подпомагайки възстановяването на разходите за грижи, осигурявайки социална и психологическа подкрепа и предоставяйки достъп до научни постижения, дори без точна диагноза. Това проучване включва над 11.6 милиона скринирани новородени за болестта на Помпе от 29 различни универсални програми за скрининг в 8 страни и 4 континента. Болестността на болестта на Помпе при раждане е 1:18,711, без доказателства за разлики между популации от европейски, латиноамерикански или азиатски произход, въпреки че могат да съществуват разлики за подтипове на болестта на Помпе. Това проучване също така сравнява тези резултати, базирани на директно откриване на заболяването и анализирани с помощта на биномен метод заедно с анализ на мощността, с други методи за оценка на ‘честотата’ на редките генетични заболявания.
Това проучване включва над 11.6 милиона скринирани новородени за болестта на Помпе от 29 различни универсални програми за скрининг в 8 страни и 4 континента. Болестността на болестта на Помпе при раждане е 1:18,711, без доказателства за разлики между популации от европейски, латиноамерикански или азиатски произход, въпреки че могат да съществуват разлики за подтипове на болестта на Помпе. Това проучване също така сравнява тези резултати, базирани на директно откриване на заболяването и анализирани с помощта на биномен метод заедно с анализ на мощността, с други методи за оценка на ‘честотата’ на редките генетични заболявания.